- α-Mannosidose
-
Klassifikation nach ICD-10 E77.1 Defekte beim Glykoproteinabbau
MannosidoseICD-10 online (WHO-Version 2011) Die α-Mannosidose ist eine sehr seltene autosomal-rezessiv vererbte lysosomale Speicherkrankheit.
Inhaltsverzeichnis
Prävalenz
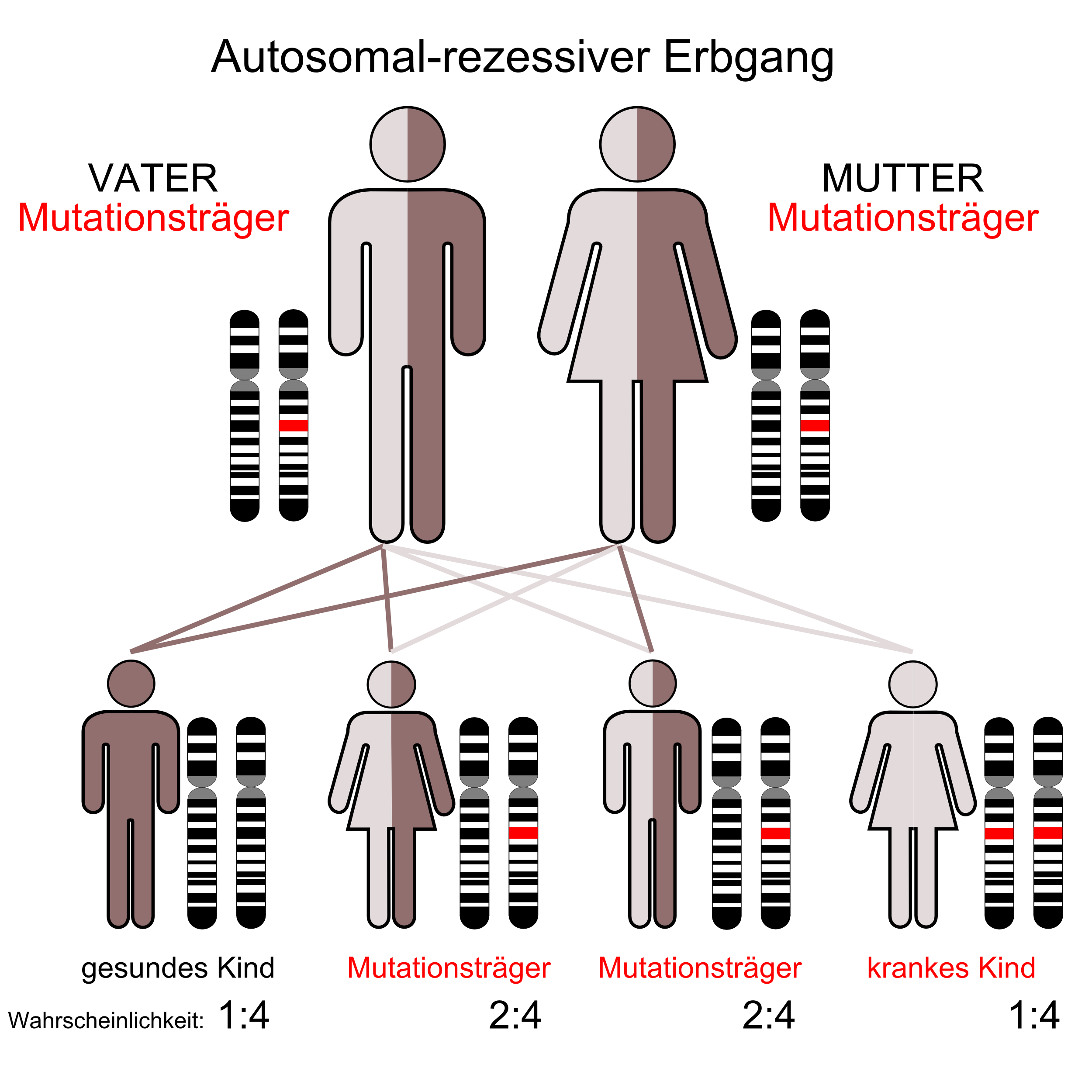
 Autosomal-rezessiver Erbgang
Autosomal-rezessiver Erbgang
Die α-Mannosidose ist mit einer Prävalenz von etwa 1 : 500.000 bei lebendgeborenen Kindern eine ausgesprochen seltene Erkrankung.[1]
Symptome
Die α-Mannosidose ist bei den betroffenen Patienten durch Immunschwäche, Anomalien des Gesichts, Veränderungen am Skelett, Schwerhörigkeit und intellektuelle Defizite gekennzeichnet. Die ersten Symptome können schon bei der Geburt sichtbar sein. Mit zunehmendem Alter schreitet die Krankheit aber voran und der Zustand der Betroffenen verschlechtert sich weiter. Neben den bereits erwähnten Symptomen können einige mit Klumpfuß geboren werden oder in den ersten zwölf Monaten einen Hydrocephalus (Wasserkopf) entwickeln.[1]
Die Immunschwäche bedingt vor allem im ersten Lebensjahrzehn rezidive Infekte. Unter den Skelettanomalien finden sich unter anderem leicht- bis mittelgradiger Dysostosis multiplex, Skoliose (Seitverbiegung der Wirbelsäule) und Deformation des Brustbeins. Die Anomalien des Gesichts sind ein vergrößerter Schädel, eine markante Stirn, abgerundete Augenbrauen, Sattelnase, Makroglossie (vergrößerte Zunge), weit auseinanderstehende Zähne und Progenie (eine Form der Kieferfehlstellung). Leichtes Schielen ist ebenfalls ein häufiges Symptom.[1]
Die Schwerhörigkeit zeichnet sich durch eine mittlere bis schwere Störung der Schallempfindung aus. Die Motorik der Patienten wird auch durch Muskelschwäche, Anomalien der Gelenke und Ataxie beeinträchtigt.[1]
Von Patient zu Patient können sich die Symptome erheblich unterscheiden.[1]
Genetik und Pathogenese
 Bändermodell des Enzyms alpha-Mannosidase.[1]
Bändermodell des Enzyms alpha-Mannosidase.[1]Der α-Mannosidose liegt ein autosomal-rezessiver Erbgang zugrunde. Mutationen im MAN2B1-Gen, das sich auf Chromosom 19 Genlocus p13.2-q12 befindet, sind die Ursache der Erkrankung. Das MAN2B1-Gen kodiert für das Enzym α-Mannosidase. Mutationen in diesem Genprodukt können eine verminderte Aktivität der α-Mannosidase bewirken, wodurch sich im Gewebe der betroffenen Patienten mannosereiche Glycokonjugate anreichern.
Diagnose
 Transmissionselektronenmikroskopische Aufnahme eines Lymphozyten. Links ein vakuolisierter Lymphozyt eines an Mannosidose erkrankten Patienten und Rechts, im Vergleich dazu, ein Lymphozyt eines Gesunden.[1]
Transmissionselektronenmikroskopische Aufnahme eines Lymphozyten. Links ein vakuolisierter Lymphozyt eines an Mannosidose erkrankten Patienten und Rechts, im Vergleich dazu, ein Lymphozyt eines Gesunden.[1]Die Diagnose kann durch die Bestimmung der Aktivität der α-Mannosidase in Leukozyten oder anderen kernhaltigen Zellen gestellt werden. Eine DNA-Analyse (‚Gentest‘) kann zur Bestätigung der Ergebnisse durchgeführt werden. Die Ausscheidung erhöhter Mengen von mannosereichen Oligosacchariden über den Urin ist ein Indiz für die Erkrankung, aber kein spezifischer Nachweis. Eine pränatale Diagnose ist sowohl biochemisch als auch molekulargenetisch möglich.[1]
Therapie
Es gibt bisher keinen etablierte kurative Therapie. Ein vielversprechender Therapieansatz ist die Enzymersatztherapie (ERT), die bisher allerdings erst in präklinischen Studien getestet werden konnte. Im Tiermodell Meerschweinchen konnte damit die Akkumulation der Oligosaccharide im Gewebe reduziert werden. Eine Ausnahme bildete dabei das Gehirn, da das applizierte Enzym, bedingt durch die Blut-Hirn-Schranke, nicht das Gehirn erreichen kann.[2] Ähnliche Versuche bei Knockout-Mäusen − hier wurde das Man2b1-Gen der Mäuse abgeschaltet − führten überraschenderweise auch zu einer Abnahme der Oligosaccharide im Gehirn der Versuchstiere.[3] Die ERT für Patienten mit α-Mannosidose ist das Ziel des europäischen Projektes Hue-Man.[4]
Bei einigen Patienten wurde eine allogene Stammzelltransplantation durchgeführt.[5][6][7][8] Die Ergebnisse waren zum Teil recht vielversprechend. Der therapeutische Nutzen ist aber bei den erheblichen Risiken der allogenen Stammzelltransplantation genau abzuwägen. Die allogene Stammzelltransplantation ist vor allem bei jungen Patienten in der ersten Lebensdekade, mit weniger stark fortgeschrittener Erkrankung, eine Therapieoption.[1]
Die Gabe von Zinksulfat (Zink-Substitution) zeigte in vitro eine signifikante Steigerung der Aktivität der αMannosidase, weshalb dies anfänglich ein verbreiteter Therapieansatz war. In Langzeitstudien zeigte sich jedoch bei Patienten mit α-Mannosidose kein signifikanter Effekt.[9]
Die sonst übliche Therapie erfolgt rein symptomatisch. Dabei wird idealerweise eine proaktive Behandlung, wie beispielsweise Krankengymnastik, die möglichen zukünftigen Komplikationen vorbeugt, angewendet. Die durch die Immunschwäche bedingten Infekte müssen häufig behandelt werden.
Prognose
Der Zustand der Patienten verschlechtert sich mit zunehmendem Alter. Die Funktion der Skelettmuskulatur und die Motorik lassen zunehmend nach, wodurch der Großteil der Betroffenen rollstuhlpflichtig wird. Kein Patient ist vollständig sozial unabhängig. Viele Patienten werden älter als 50 Jahre. Mit zunehmender Progression der Erkrankung werden alle Patienten schwerhörig und benötigen ein Hörgerät.[1]
Erstbeschreibung
Der Arzt Per-Arne Öckerman (* 1933) von der Universität Lund in Schweden beschrieb 1967 als Erster bei einem Jungen eine neue Form einer lysosomalen Speicherkrankheit mit ähnlichen Symptomen wie das Hurler-Syndrom, bei der aber keine Mukopolysaccharide akkumuliert werden.[10]
Veterinärmedizin
Bei Rindern, insbesondere bei der Rasse ‚Aberdeen Angus‘ ist die α-Mannosidose eine relativ häufig verbreitete Krankheit.[11][12]
Weiterführende Literatur
- K. W. Moremen: Golgi alpha-mannosidase II deficiency in vertebrate systems: implications for asparagine-linked oligosaccharide processing in mammals. In: Biochim Biophys Acta 1573, 2002, S. 225–235. PMID 12417404
- T. Beccari u. a.: Lysosomal alpha-D-mannosidase. In: Biosci Rep 19, 1999, S. 157–162. PMID 10513892
- J. P. Kistler u. a. Mannosidosis: new clinical presentation, enzyme studies, and carbohydrate analysis. In: Arch Neurol 34, 1977, S. 45–51. PMID 12732
- Y. Gotoda u. a.: Missense and nonsense mutations in the lysosomal alpha-mannosidase gene (MANB) in severe and mild forms of alpha-mannosidosis. In: Am J Hum Genet 63, 1998, S. 1015−1024. PMID 9758606
- A. Gutschalk u. a.: Adult alpha-mannosidosis: clinical progression in the absence of demyelination. In: Neurology 63, 2004, S. 1744–1746. PMID 15534274
Weblinks
- Α-Mannosidose bei Online Mendelian Inheritance in Man
- Α-Mannosidose bei Orphanet (Datenbank für seltene Krankheiten)
Einzelnachweise
- ↑ a b c d e f g h i j k D. Malm und O. Nilsson: Alpha-mannosidosis. In: Orphanet Journal of Rare Diseases 3, 2008, 21. doi:10.1186/1750-1172-3-212008 (Review, Open Access, CC-by-2.0)
- ↑ A. C. Crawley u. a.: Enzyme replacement therapy in alpha-mannosidosis guinea-pigs. In: Mol Genet Metab 89, 2006, S. 48−57. PMID 16807033
- ↑ D. P. Roces u. a.: Efficacy of enzyme replacement therapy in alpha-mannosidosis mice: a preclinical animal study. In: Hum Mol Genet 13, 2004, S. 1979−1988. PMID 15269179
- ↑ Towards The Development Of An Effective Enzyme Replacement Therapy For Human Alpha-Mannosidosis. Abgerufen am 7. November 2009
- ↑ S. S. Grewal u. a.: Effective treatment of alpha-mannosidosis by allogeneic hematopoietic stem cell transplantation. In: J Pediatr 144, 2004, S. 569−573. PMID 15126988
- ↑ D. A. Wall u. a.: Bone marrow transplantation for the treatment of alpha-mannosidosis. In: J Pediatr 133, 1998, S. 282−285. PMID 9709723
- ↑ A. Will u. a.: Bone marrow transplantation in the treatment of alpha-mannosidosis. In: Arch Dis Child 62, 1987, S. 1044−1049. PMID 3314721; PMC 1778651
- ↑ S. U. Walkley u. a.: Bone marrow transplantation corrects the enzyme defect in neurons of the central nervous system in a lysosomal storage disease. In: PNAS 91, 1994, S. 2970−2974. PMID 8159689; PMC 43496
- ↑ L. T. Wong u. a.: Oral zinc therapy in the treatment of alpha-mannosidosis. In: Am J Med Genet 46, 1993, S. 410−414. PMID 8357013
- ↑ P. A. Öckerman: A generalised storage disorder resembling Hurler's syndrome. In: The Lancet 2, 1967, S. 239.
- ↑ J. D. Hocking u. a.: Deficiency of alpha-mannosidase in Angus cattle: an inherited lysosomal storage disease. In: Biochem J 128, 1972, S. 69–78. PMID 4673577; PMC 1173571
- ↑ H. W. Leipold u. a.: Mannosidosis of Angus calves. In: J Am Vet Med Assoc 175, 1979, S. 457–459. PMID 500478

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Lysosomale Speicherkrankheit
- Krankheitsbild in der Kinderheilkunde
- Zellbiologie



Wikimedia Foundation.