- Chorea major
-
Klassifikation nach ICD-10 G10 Chorea Huntington
Chorea chronica progressiva hereditariaF02.2* Demenz bei Chorea Huntington ICD-10 online (WHO-Version 2006) 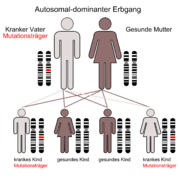 Der autosomal-dominante Erbgang
Der autosomal-dominante ErbgangDie Chorea major (Huntington) auch als Huntington-Chorea bezeichnet (älterer Name: Veitstanz) ist eine bis heute unheilbare vererbliche Erkrankung des Nervensystems.
Es ist eine autosomal-dominant vererbte, neuro-degenerative Erkrankung, die meist zwischen dem 30. und 60. Lebensjahr zu ersten Krankheitssymptomen führt. Diese sind Bewegungsstörungen und psychische Symptome. Männer und Frauen sind in gleicher Häufigkeit betroffen. Die Krankheit nimmt immer einen schweren Verlauf und führt im Durchschnitt 15 Jahre nach den ersten Symptomen zum Tod. Homozygote (gleicherbige) Mutationsträger, d. h. solche mit zwei mutierten Allelen (Gen-Strängen) sind nicht stärker betroffen als heterozygote (verschiedenerbige) und alle Merkmalsträger erkranken früher oder später (vollständige Penetranz). Die Chorea Huntington ist eine der häufigsten erblich bedingten Hirnstörungen mit einer Inzidenz von 5:100.000, diese schwankt jedoch von Land zu Land erheblich (Bsp. Japan: 1:100.000). Seit 1993 lässt sich das krankmachende Allel auf dem kurzen Arm des vierten Chromosoms (Locus 4p16.3, [1]) nachweisen, auch beim Ungeborenen durch Amniozentese oder Chorionzottenbiopsie.
Inhaltsverzeichnis
Herkunft des Namens
Die Chorea Huntington (chorea, gr. χορεία = Tanz) wurde 1872 von dem New Yorker Arzt George Huntington ausführlich beschrieben. Er erkannte, dass diese Besonderheit in manchen Fällen vererbt wird. Er war jedoch anfangs der fälschlichen Annahme, dass die Ausbreitung von Chorea H. auf Long Island (USA) beschränkt sei. Tatsächlich war sie aber bereits damals weltweit anzutreffen. Der deutsche Name ist „erblicher Veitstanz“. Die Bezeichnung Veitstanz ist seit dem 16. Jh. bezeugt und hat ihren Ursprung darin, dass als Helfer der heilige Veit (Vitus), angerufen wurde. Wieso gerade dieser Heilige angerufen wurde, ist nicht bekannt. Wegen beim europäischen Gerichtshof vorliegender Beschwerden und gerichtlicher Urteile ist der Name „Veitstanz“ mittlerweile nicht mehr gebräuchlich, da er in Verbindung mit der negativen Eugenik der Nationalsozialisten gebracht wird.[2] Mittlerweile ist in allen ärztlichen und sonstigen Fachkreisen der Begriff „Chorea Huntington“ als Bezeichnung üblich.
Pathophysiologie
Genetik
Chorea Huntington ist eine einfach autosomal dominant vererbte Krankheit. Dies bedeutet, dass die Nachkommen eines Betroffenen mit einer Wahrscheinlichkeit von mindestens 50 % ebenfalls betroffen sein können – je nachdem, ob das phänotypisch erkrankte Elternteil ein oder zwei mutierte Allele besitzt (zwei mutierte Allele = 100 % Wahrscheinlichkeit für eine Erkrankung). Generationensprünge kommen nicht vor, Männer und Frauen sind gleich häufig betroffen. Davon abweichend liegt bei ca. 5 bis 10 % der Patienten eine Neumutation vor. Das Protein, das die Krankheit verursacht, heißt Huntingtin, das dafür codierende Gen liegt auf dem kurzen Arm von Chromosom 4 (Locus 4p16.3). Bei gesunden Menschen wiederholt sich dort das Basentriplett CAG circa 9 bis 35 Mal. Bei Kranken kommt dieses Triplett von 36 bis zu 250 Mal vor. Dies wird vor allem durch sogenannte „slippage“ (ein Verrutschen der DNA-Polymerase bei der Replikation) oder (unwahrscheinlicher, aber immerhin möglich) durch ein nichtreziprokes (asymmetrisches) Crossing-over verursacht. Je häufiger sich diese Wiederholung ereignet, desto früher tritt die Erkrankung auf (Antizipationseffekt). Die juvenile Chorea H. manifestiert sich bei über 60 CAG-Tripletts. Ein Ausbruch im vierten Lebensjahr ist beschrieben. Bei Vererbung durch den Vater erhöht sich die Zahl der CAG-Tripletts häufiger als bei Vererbung durch die Mutter (Imprinting-Phänomen). Meist findet sich bei einem Elternteil eines Betroffenen mit Neumutation schon eine Anzahl von 30 bis 35 Wiederholungen (Prämutation).
Molekularbiologie
Die aus dem Triplett CAG resultierende mRNA codiert für die Aminosäure Glutamin. Das mutierte Huntingtin besitzt also mehr als die übliche Anzahl an aneinandergereihten Glutamin-Resten. Möglicherweise handelt es sich dabei um eine „Gain-of-Function-Mutation“, das heißt die normale Funktion des Huntingtin-Proteins könnte erhalten bleiben, zusätzlich jedoch erhält es weitere – toxische – Eigenschaften. Eine hohe Expression von Huntingtin führt zu amyloidähnlichen Ablagerungen (inclusions) von mutiertem Huntingtin, wahrscheinlich auch deshalb, weil der Abbau des mutierten Proteins durch das Proteasom nicht mehr richtig funktioniert. Andererseits wurde auch eine Toxizität des freien mutierten Huntingtins von einigen Arbeitsgruppen nachgewiesen, so dass die Huntingtin-Aggregate als Schutz angesehen werden können. Die betroffenen Zellen haben einen gestörten Glukosemetabolismus. Dies führt zu einer gesteigerten Empfindlichkeit gegenüber oxidativem Stress und dem erregenden Neurotransmitter Glutamat. Die betroffenen Zellen besitzen besonders viele Glutamatrezeptoren und haben viele eingehende glutamaterge Verbindungen. Trotzdem ist derzeit nur unbefriedigend erklärbar, warum die Toxizität nur in den beschriebenen Arealen nachweisbar ist, obwohl Huntingtin in allen kernhaltigen Zellen gebildet wird.
Die physiologische Funktion von Huntingtin ist trotz intensiver Forschung noch nicht geklärt. Interessanterweise scheint es funktionelle Ähnlichkeiten mit Ataxin-2 zu besitzen. Ataxin-2 ist ein Protein welches eine andere Polyglutaminerkrankung auslöst, nämlich die Spinozerebelläre Ataxie Typ 2.
Neuroanatomie und Physiologie
Betroffen ist das Putamen, welches ein Teil des Corpus striatum in den Basalganglien ist und über einen direkten und über einen indirekten Pfad den Globus pallidus internus hemmen kann. Bei Menschen mit Chorea Huntington degenerieren GABA/Enkephalin-erge Neurone, d. h. der Anfang des indirekten Pfades ist zerstört. Dies hat zur Folge, dass der Globus pallidus internus noch stärker gehemmt wird als bei gesunden Menschen. Da der Globus pallidus internus seinerseits normalerweise den Thalamus inhibiert, wird dieser nun weniger gehemmt, also aktiviert (=Disinhibition). Die Konsequenz ist eine Übererregung des Thalamus und des Cortex.[3]
Da die indirekten Verbindungen im Verlauf meist zuerst zerstört werden, steht am Anfang der Erkrankung eine Überaktivierung mit überschießenden Bewegungen im Vordergrund. Im weiteren Verlauf gehen auch die direkten Verbindungen verloren und es dominieren Bewegungsarmut (Akinese) und Steifheit (Rigor).
Krankheitsbild
Verlauf
Die Krankheit bleibt meist lange Zeit stumm, bis sie zwischen dem 30. und 40. Lebensjahr ausbricht. Generell sind aber Ausbrüche zwischen dem 3. und dem 75. Lebensjahr beschrieben. Patienten mit einem frühen Krankheitsbeginn zeigen häufig einen schwereren Verlauf. Psychische Beschwerden gehen den Bewegungsstörungen häufig und oft viele Jahre voraus. Die Bewegungsstörungen beginnen meist mit Hyperkinesien (ungewollten Bewegungen) bei verringertem Muskeltonus. Später zeigen sich eher Hypokinesie (Bewegungsarmut) und Erhöhung des Muskeltonus. Eine Verlaufsform, bei der die Bewegungsarmut von Anfang an im Vordergrund steht, wird Westphal-Variante genannt und tritt häufiger bei frühem Krankheitseintritt auf. Die Chorea Huntington nimmt einen über bis zu 15 bis 20 Jahre dauernden Verlauf, der mit dem Tod endet. Das Voranschreiten der Krankheit kann durch zu hohen Stress beschleunigt werden, umgekehrt haben günstige Lebensumstände mit einer leidensgerechten Aktivierung der Betroffenen einen günstigen Einfluss auf den Verlauf der Erkrankung.
Psychische Beschwerden
Zu den ersten Erscheinungen der psychischen Veränderung gehören meist Störungen des Affektes und des Antriebes. Später können ein unbedachtes und impulsives Verhalten sowie eine Enthemmung in zwischenmenschlichen Beziehungen auftreten. Aufgrund der mangelhaften Kontrolle über die Muskulatur (z. B. des Gesichtes mit Grimassieren) kann der falsche Eindruck eines bereits fortgeschrittenen Persönlichkeitsverlustes entstehen, was bei den Patienten Resignation und Depressionen hervorruft. Besonders in der Frühphase der Erkrankung kann dies zu suizidalem Verhalten führen. Früh treten auch Störungen der visuellen Informationsverarbeitung auf, was z. B. dazu führt, dass die Kranken insbesondere kritische Gesichtsausdrücke ihrer Mitmenschen – wie z. B. Verärgerung – nicht richtig wahrnehmen und so darauf nicht angemessen reagieren können. Im Frühstadium werden leichte Beeinträchtigungen der intellektuellen Fähigkeiten sowie Gedächtnisstörungen oft übersehen. Im Spätstadium der Erkrankung entwickeln die Patienten eine Demenz, d. h. es kommt zum Verlust ihrer kognitiven Fähigkeiten. So finden sich Störungen der Merkfähigkeit, damit im Zusammenhang stehend eine Desorientierung und eine Sprachverarmung. Einige Patienten entwickeln Wahnvorstellungen, die dazu führen, dass sie als schizophrene Patienten in psychiatrischen Kliniken behandelt werden (psychisch betonter Verlauf).
Bewegungsstörungen
Die Chorea beginnt meist mit einer zunächst kaum bemerkbaren Bewegungsunruhe der Arme und Beine, des Gesichtes, später des Kopfes sowie des Rumpfes. Diese Unruhe kann sich zu heftigen choreatischen Hyperkinesien steigern. Das sind plötzlich einsetzende, unwillkürliche Bewegungen verschiedener Muskeln, wodurch die Willkürbewegungen unterbrochen werden. Betroffene versuchen zunächst, die choreatischen Bewegungen zu verbergen, indem sie diese in willkürliche Bewegungsabläufe einbauen, z. B. streichen sich nach einer einschießenden Beugebewegung des Armes über das Haar. Zunehmend geraten die Muskelbewegungen aber außer Kontrolle. Beim Vollbild der Erkrankung kommt es zum plötzlichen Grimassieren und zu schleudernden Bewegungen (Ballismus) von Armen und Beinen. Sprechen und Schlucken fallen zunehmend schwer (Dysarthrie und Schluckstörung). Typischerweise beginnen diese Hyperkinesien in den rumpffernen Teilen der Extremitäten (in den Händen und im Gesicht), so wird der Mund weit geöffnet, die Zunge weit rausgestreckt und sofort wieder zurückgezogen („Chamäleonzunge“). Im weiteren Verlauf sind auch die rumpfnahen Extremitätenanteile betroffen. Bei Auslösen des Kniesehnenreflexes bleibt das Knie gestreckt (Gordon-Phänomen). Die Bewegungsunruhe verstärkt sich unter seelischer und körperlicher Belastung. Obwohl die unkontrollierten Bewegungen im Schlaf aufhören, nehmen sie bei Ermüdung eher zu. Die anfangs choreatischen Hyperkinesien wandeln sich mit zunehmendem Krankheitsverlauf in Dystonien, wobei durch Erhöhung der Muskelspannung (Muskeltonus) die Gliedmaßen minuten- bis stundenlang in einer manchmal schmerzhaften Fehlstellung verharren. An Stelle des Grimassierens tritt dann eventuell eine Anarthrie auf, d. h. es kann eine völlige Unfähigkeit bestehen, Sprechbewegungen auszuführen und der Patient ist nicht mehr in der Lage, durch Mimik, Gestik und Sprache zu reagieren. Das Schlucken fällt den Patienten immer schwerer und kann zu lebensbedrohlichen Komplikationen führen, zumal die Patienten durch die Hyperkinesien einen erhöhten Energieverbrauch haben. Dieser kann sich im Endstadium der Erkrankung auf mehr als das Fünffache des normalen Grundumsatzes erhöhen, so dass eine adäquate Versorgung nur noch mit ergänzender parenteraler Nahrungszufuhr möglich ist.
Diagnostik
Die Diagnose kann meist klinisch anhand der Symptome gestellt werden. Darüber hinaus besteht die Möglichkeit, die Diagnose durch genetische Analyse zu sichern, auch schon bevor sich die ersten Symptome zeigen, sogar vor der Geburt. Weitere Möglichkeiten sind eine Kernspintomographie oder Computertomographie. Sie zeigen eine Atrophie des Corpus striatum und hier vor allem des Nucleus caudatus. Diese Atrophie führt zu einer Erweiterung der Seitenventrikel. In der 18FDG-PET zeigt sich eine Störung des Glukosestoffwechsels im Corpus striatum. Eine mögliche Differentialdiagnose ist CJD (Creutzfeldt-Jakob-Krankheit) sowie vCJD (variant CJD).
Ethische Probleme der humangenetischen Diagnostik
Es ist heute möglich, weit vor dem Auftreten jeglicher Symptome bei Menschen aus betroffenen Familien eindeutig festzustellen, ob sie den zur Chorea Huntington führenden Gendefekt haben oder nicht. Während für Kinder eines betroffenen Elternteils ohne weitere Informationen die Wahrscheinlichkeit des Auftretens der Erkrankung bei 50 % liegt, ist nach einer solchen Diagnostik eindeutig geklärt, ob der betreffende Mensch entweder niemals oder aber mit Sicherheit die Erkrankung bekommen wird. Die Entscheidung darüber, ob eine solche Diagnostik gewünscht wird, ist höchst persönlich und kann nur nach einer umfassenden Aufklärung getroffen werden. Zugleich muss berücksichtigt werden, dass mit einer solchen Diagnostik auch Informationen über andere Blutsverwandte bekannt werden. Jemand mit einem choreakranken Großelternteil, der positiv auf die Chorea-Genveränderung getestet wird, weiß damit genau, dass auch der entsprechende Elternteil Träger dieser Genveränderung ist, auch wenn dieser derzeit noch keinerlei Krankheitssymptome hat.
Therapie
Eine Therapie, welche die Krankheit an sich heilt oder dauerhaft aufhält, ist nicht bekannt. Verschiedene Vitamine und Nahrungsergänzungsmittel werden mit unterschiedlichem Erfolg eingesetzt, um die Zellen vor oxidativem Stress zu schützen und so den Krankheitsverlauf zu verlangsamen. Das Medikament Riluzol vermindert die Glutamatausschüttung und soll den Verlauf verlangsamen. Sämtliche Therapien werden flankiert von physiotherapeutischer, ergotherapeutischer und logopädischer Behandlung zur Besserung der Bewegungsfähigkeit beziehungsweise der Sprache und Schluckfähigkeit. Gleichzeitig sollten der Patient und auch seine Angehörigen psychologisch betreut werden. Diese drei Behandlungen bilden die Grundlage jeder medikamentösen Therapie. Die Ernährung sollte den erhöhten Energiebedarf, die Schluckbeschwerden und den erhöhten Zuckerbedarf der Patienten berücksichtigen. Einzelne Symptome können ebenfalls je nach Bedarf behandelt werden.
Behandlung der Bewegungsstörungen
Gegen Hyperkinesien werden neben Tetrabenazin auch Dopamin-Antagonisten eingesetzt, meist Tiaprid oder auch Sulpirid. Bei einsetzendem Rigor werden Dopamin-Agonisten oder L-Dopa eingesetzt, können jedoch die Hyperkinesien verstärken. Daher wird die medikamentöse Therapie erst eingeleitet, wenn die Bewegungsstörungen den Patienten im Alltag stark beeinträchtigen.
Behandlung psychischer Symptome
Bei psychotischen Symptomen wird auf atypische Neuroleptika zurückgegriffen, gegen depressive Symptome vor allem Antidepressiva aus der Gruppe der SSRIs. Bei Schlafstörungen und Angstzuständen kommen Benzodiazepine zum Einsatz.
Neuroprotektive Behandlung
Es gibt Studien, die eine neuroprotektive Wirkung von Gabapentin auf Patienten mit Chorea Huntington implizieren. Durch dieses Medikament soll die Excitotoxizität des Glutamats auf die Nervenzelle reduziert werden.[4]
Einzelnachweise
- ↑ Darstellung des Gens im NCBI Map Viewer
- ↑ Hess V.:Vorlesung Geschichte Theorie Ethik: Medizin im Nationalsozialismus, Institut für Geschichte der Medizin, Charité – Hochschulmedizin Berlin
- ↑ Mahlon R. DeLong: The Basal Ganglia. In: Eric R. Kandel, James H. Schwartz und Thomas M.Jessell:Principles of Neural Science. 2000, S. 860.
- ↑ J. O. Heidenreich u. a.,1H-MRS zur Untersuchung der neuroprotektiven Wirkung von Gabapentin bei Chorea Huntington
Literatur
- J. Rutishauser: Morbus Huntington: disrupt the fatal attraction. In: Schweiz Med Forum, 24/2002, S. 586–587.
- E. Cattaneo u. a: Das Rätsel der Chorea Huntington In: Spektrum der Wissenschaft, JANUAR 2004, S. 60–66.
Weblinks
- Leitlinie Chorea der Deutschen Gesellschaft für Neurologie bei AWMF online (Stand 10/2005)
- Links zum Thema Chorea Huntington im Open Directory Project
Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.