- Metallcharakter
-
Die chemische Bindung ist ein physikalisches Phänomen, durch das zwei oder mehrere Atome oder Ionen fest zu chemischen Verbindungen aneinander gebunden sind. Dieses beruht darauf, dass es für die meisten Atome oder Ionen energetisch günstiger ist, an geeignete Bindungspartner gebunden zu sein, anstatt freie einzelne Teilchen zu bilden. Grundlage der Bindung sind entweder elektrostatische Wechselwirkungen oder Wechselwirkungen der Elektronen zweier oder mehrerer Atome. In vielen Fällen spielen beide Bindungsarten eine Rolle. Parameter, die zur Beschreibung einer Bindung wichtig sind und sich experimentell untersuchen lassen, sind die Bindungslänge als Maß für den Abstand zweier Atome und die Bindungsenergie, die die Stärke einer Bindung angibt. Die chemische Bindung ist die Grundlage dafür, dass sich Moleküle und damit überhaupt chemische Verbindungen bilden können und ist somit eine der wichtigsten Grundlagen der Chemie.
Chemische Bindungen können je nach Art der Bindung in verschiedene Typen eingeteilt werden. In Ionenkristallen herrscht die auf elektrostatischen Wechselwirkungen beruhende Ionenbindung vor, in Metallen die auf frei bewegliche Elektronen beruhende metallische Bindung. In Molekülen und Komplexen liegen dagegen lokalisierte Bindungen vor, die auf der Bildung von Elektronenpaaren beruhen. Innerhalb der lokalisierten Elektronenpaarbindungen wird häufig zwischen der kovalenten Bindung, bei der jedes Atom ein Elektron zur Bindung beiträgt, und der dativen Bindung in Komplexen, bei der ein Elektronenpaar eines Liganden mit einem leeren Orbital des Zentralatoms wechselwirkt, unterschieden. In speziellen Fällen können Mehrzentrenbindungen auftreten.
Mitunter werden schwache Wechselwirkungen, wie die Van-der-Waals-Wechselwirkungen, Dipol-Wechselwirkungen und die Wasserstoffbrückenbindung zu den Bindungen gezählt. Jedoch sind diese keine festen chemischen Bindungen, sondern schwache Anziehungskräfte, die zwischen einzelnen Molekülen wirken.
Bindungen lassen sich durch die Zuführung von Energie, etwa in Form von Wärme oder Licht, spalten. Die Atome oder Molekülreste, die an die gespaltene Bindung grenzen, haben häufig eine hohe Neigung, wieder eine Bindung zu bilden. Die Neubildung der Bindung kann nicht nur an der vorher gespaltenen Stelle stattfinden, sondern auch mit anderen Atomen oder Molekülen erfolgen. Dies ist die Grundlage für chemische Reaktionen.
Inhaltsverzeichnis
Geschichte
 Linus Pauling erhielt 1954 den Chemie-Nobelpreis unter anderem für seine Arbeiten zur chemischen Bindung
Linus Pauling erhielt 1954 den Chemie-Nobelpreis unter anderem für seine Arbeiten zur chemischen BindungDie Entwicklung von verschiedenen Theorien zur chemischen Bindung ist eng mit der Entwicklung von Theorien und Experimenten zur Gestalt des einzelnen Atoms verbunden. Die ersten konkreten Theorien wurden nach der Entdeckung des Elektrons durch Joseph John Thomson 1897 aufgestellt. In seinem Atommodell stellte Thomson sich vor, dass die chemischen Bindungen auf elektrostatischen Kräften beruhten, die durch den Transfer von einem zum anderen Atom entstanden. Dadurch ergab sich zunächst die Annahme, dass chemische Bindungen immer polar aufgebaut sein müssen.[1]
Auf Grund der Eigenschaften organischer Verbindungen, die nicht mit polaren Bindungen zu erklären waren und Versuchen mit Kanalstrahlen wurde bald klar, dass es auch eine unpolare Bindung geben muss. Gilbert Lewis vermutete erstmals 1916, dass die unpolare Bindung auf gepaarte Elektronen beruht. Diese Theorie war auch mit den Atommodellen von Rutherford und Bohr vereinbar, die währenddessen das Thomsonsche Modell abgelöst hatten.[1]
Mit der Entwicklung der Quantenmechanik und vor allem der Aufstellung der Schrödingergleichung durch Erwin Schrödinger 1926 konnten genauere Theorien der Bindung aufgestellt werden. Die erste quantenmechanische Theorie wurde mit der Valenzstrukturtheorie 1927 durch Walter Heitler und Fritz London entwickelt.[2] Die ursprüngliche Theorie war zunächst nur für das einfachste Molekül, das H2+-Ion aus zwei Protonen und einem Elektron gültig. Linus Pauling erweiterte die Theorie durch die Einführung des Orbitals und der Hybridisierung umfangreich, so dass die Theorie auf kompliziertere Moleküle angewendet werden konnte.[1]
Ebenfalls im Jahr 1927 wurde von Friedrich Hund und Robert Mulliken die genauere Molekülorbitaltheorie aufgestellt. Auch diese war zunächst nur für einfache Moleküle anwendbar, wurde jedoch nach und nach, beispielsweise durch eine genauere Erklärung von Mehrfachbindungen mit der Erklärung der π-Bindung durch Erich Hückel 1930, erweitert.[2]
Ionische Bindung
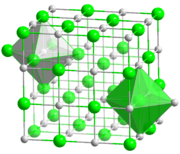 Stilisiertes Ionengitter (Natriumchlorid-Struktur)
Stilisiertes Ionengitter (Natriumchlorid-Struktur)- Hauptartikel: Ionische Bindung
Die ionische Bindung ist eine langreichweitige, ungerichtete Bindung, die in allen Raumrichtungen gleich stark wirkt. Sie ist die vorherrschende Bindungsart bei Salzen, also Verbindungen von Metallen und Nichtmetallen, die periodisch angeordnet sind. Bei der Reaktion dieser kommt es durch die große Elektronegativitätsdifferenz zu einer Übertragung von Valenzelektronen des Metalls auf das Nichtmetall und damit zu geladenen Atomen, den sogenannten Ionen. Je stärker die Elektronegativitätsdifferenz ist, desto stärker werden die Valenzelektronen übertragen und desto ionischer ist die Bindung. Jedoch sind bei allen ionischen Bindungen auch kovalente Anteile an der Bindung vorhanden. Bei schwachen Differenzen kommt es nur zu einer geringen Übertragung und es ist für die Beschreibung der Bindung nötig, beide Anteile zu berücksichtigen.
Für die Bindung in Ionenkristallen sind vor allem elektrostatische Wechselwirkungen zwischen den verschieden geladenen Ionen verantwortlich. Die energetische Struktur lässt sich theoretisch gut mit der Gitterenergie beschreiben. Dazu werden vor allem die anziehenden und abstoßenden Kräfte zwischen den Ionen, sowie die Abstoßung der sich durchdringenden Elektronenhüllen einbezogen und das Coulomb-Gesetz aufgestellt. Auch die Art des Gitters wird über die Madelung-Konstante mit einbezogen.
Die ionische Bindung ist eine starke Bindung. Typische Werte für Gitterenergien ionischer Stoffe liegen bei 787 kJ/mol für Natriumchlorid und 3850 kJ/mol für das höher geladene Magnesiumoxid (bestimmt über den Born-Haber-Kreisprozess).[3] Dies bedingt die hohen Schmelzpunkte vieler ionisch aufgebauter Substanzen. Da die Bindung ungerichtet ist, ist sie jedoch nicht stärker als viele kovalente Bindungen. Die elektrostatische Natur der Ionenbindung bedingt die Sprödigkeit vieler Ionenkristalle, da bei Verschiebungen zwischen den Ionen leicht gleichgeladene Ionen aneinandergrenzen, die sich abstoßen und so den Kristall auseinandersprengen.
Kovalente Bindung
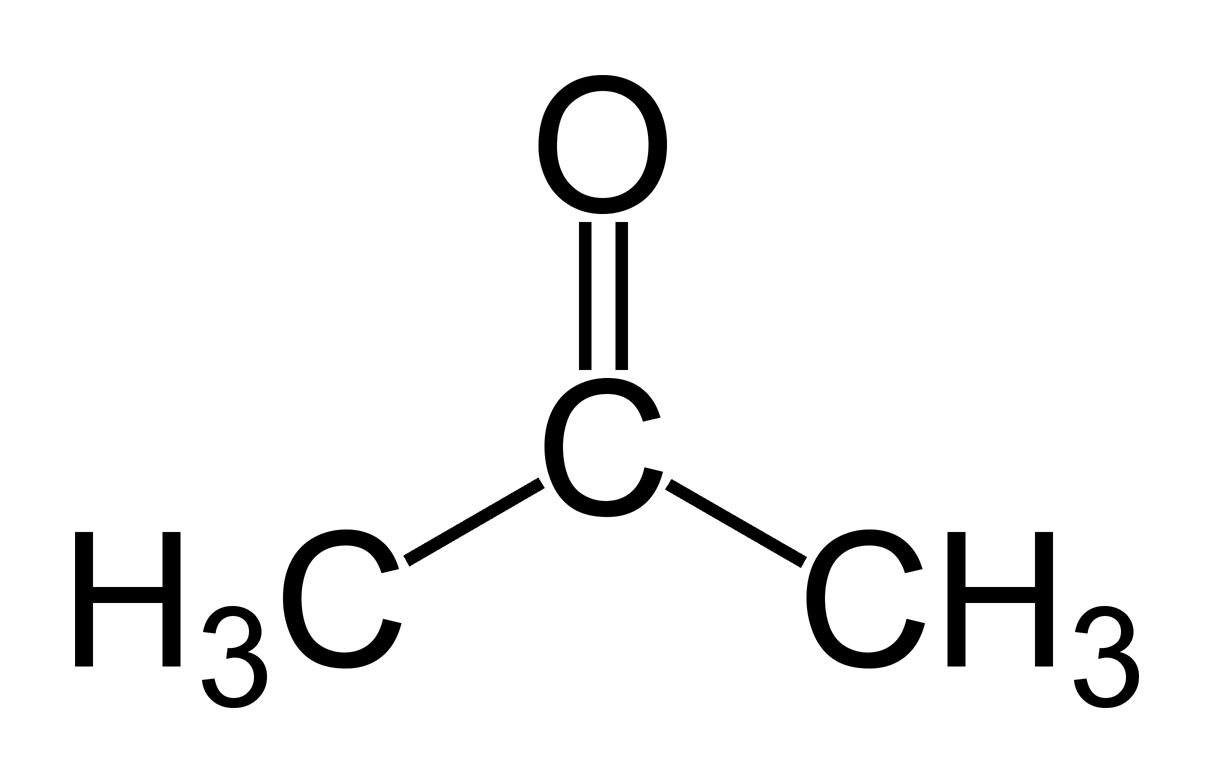
 typische Lewis-Formel (hier Aceton)
typische Lewis-Formel (hier Aceton)- Hauptartikel: Kovalente Bindung
Die kovalente Bindung ist diejenige Bindung, die in Nichtmetallverbindungen sowie Komplexen vorherrscht. Sie ist im Gegensatz zur ionischen Bindung gerichtet und an eine bestimmte Stelle zwischen zwei einzelne Atome gebunden. Ausnahme sind die Mehrzentrenbindungen, bei denen drei oder mehr Atome kovalent gebunden sind und die delokalisierten π-Bindungen, bei der mehrere Bindungen zu einer einzigen Elektronenwolke verschmolzen sind.
Kovalente Bindungen beruhen normalerweise auf einem sogenannten Elektronenpaar, das aus den Valenzelektronen der beteiligten Atome gebildet wird. Um eine kovalente Bindung in einer chemischen Formel darzustellen, wird diese in der Lewis-Formel durch einen waagerechten Strich, manchmal auch durch zwei Punkte symbolisiert. Theoretisch wird die kovalente Bindung mit zwei verschiedenen Theorien, der Molekülorbital- und der Valenzstrukturtheorie, erklärt. Ältere Theorien für Komplexe sind die Kristallfeld- und Ligandenfeldtheorie, jedoch lassen sich die Bindungsverhältnisse in Komplexverbindungen genauer durch die Molekülorbitaltheorie vorhersagen.[4]
Die Stärke einer kovalenten Bindung hängt von der Art der Bindung, den beteiligten Atomen und der Bindungslänge ab. Die stärksten kovalenten Bindungen sind die kurzen Dreifachbindungen von Elementen der zweiten Periode wie Kohlenstoff, Stickstoff oder Sauerstoff, so beträgt die Dissoziationsenergie einer Stickstoff-Stickstoff-Dreifachbindung 941,7 kJ/mol[5]. Bindungsenergien für Einfachbindungen liegen in der Regel zwischen 150 und 500 kJ/mol, bei Doppelbindungen liegen Bindungsenergien typischerweise bei 500-800 kJ/mol für die zweite Periode (O-O-Doppelbindung: 493,6 kJ/mol, C-O-Doppelbindung: 798,9 kJ/mol), für die schwächeren Doppelbindungen der höheren Perioden darunter.[5]
Valenzstrukturtheorie
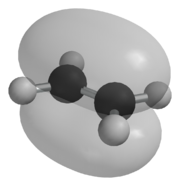 π-Bindung bei Ethen
π-Bindung bei Ethen- Hauptartikel: Valenzstrukturtheorie
Die Valenzstrukturtheorie versucht, eine Bindung quantenmechanisch zu beschreiben. Sie ist zunächst für das Wasserstoffmolekül mit zwei Protonen und zwei Elektronen mit unterschiedlichem Spin aufgestellt worden und kann durch Ergänzungen und Vereinfachungen auch für kompliziertere Moleküle angewendet werden. Für genaue Berechnungen müssen zunächst die Wellenfunktionen der beteiligten Elektronen aufgestellt werden. Diese unterscheiden sich je nach Orbital, in dem sich das Elektron befindet. Im Gegensatz zur Molekülorbitaltheorie werden die Bindungen in der Regel einzeln und nicht das Molekül als Ganzes betrachtet.
In der einfachsten Nährung wird die Gesamtwellenfunktion Ψ des H2-Moleküls als Produkt der beiden Wellenfunktionen der beiden Elektronen gesehen.
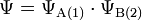
- A: 1. Atom, B: 2. Atom, 1: 1. Elektron, 2: 2. Elektron
Dies berücksichtigt noch keine Beeinflussungen der Elektronen untereinander und gilt somit exakt nur für zwei isolierte Wasserstoffatome. Für genauere Ergebnisse für gebundene Atome muss vor allem die Austauschenergie mit einbezogen werden, die dadurch zu Stande kommt, dass die Elektronen nicht an einem Atom lokalisiert sind, sondern an beiden eine Aufenthaltswahrscheinlichkeit besitzen. Die Wellenfunktion wird dann als
geschrieben. Mit weiteren Verfeinerungen, etwa der Einbeziehung der Abschirmung, kann eine weitere Annäherung der Theorie an den experimentellen Wert erreicht werden.
Ein wichtiges Konzept, mit dem die Valenzstrukturtheorie auf kompliziertere Moleküle ausweiten kann, ist die Hybridisierung. Dazu werden aus unterschiedlichen Orbitalarten Hybridorbitale gebildet. Am Bekanntesten sind die aus s- und p-Orbitalen gebildeten sp3-Hybridorbitale, die vor allem zur Erklärung der Bindungssituation in Kohlenstoffatomen verwendet werden. Dabei werden aus dem im Kohlenstoffatom vorhandenen drei p-Orbitalem und dem 2s-Orbital vier gleiche sp3-Hybridorbitale gebildet, die tetraedrisch angeordnet sind und Bindungen zu benachbarten Atomen bilden. Es ist auch möglich, dass nur eines oder zwei der p-Orbitale an der Hybridisierung beteiligt sind. Dann bilden sich sp- oder sp2-Hybridorbitale.
Je nachdem, welche Orbitale beteiligt sind, werden verschiedene Bindungsarten unterschieden. Sind an einer Bindung nur s- und/oder sp-Hybridorbitale beteiligt, handelt es sich um eine sogenannte σ-Bindung. Diese liegt direkt auf der Verbindungsachse zwischen den Atomen und besitzt keine Knotenebene. p-Orbitale bilden π-Bindungen, die oberhalb und unterhalb der Kernverbindungsachse liegen und eine Knotenebene besitzen. Sie sind für die Beschreibung von Mehrfachbindungen, vor allem der Doppel- und Dreifachbindungen wichtig. Sehr selten kommen auch Vier- oder Fünffachbindungen vor, die neben σ- und π-Bindungen aus d-Orbitalen gebildete δ-Bindungen aufweisen.
Molekülorbitaltheorie
 Molekülorbitalschema von Fluor F2
Molekülorbitalschema von Fluor F2- Hauptartikel: Molekülorbitaltheorie
Wie die Valenzstrukturtheorie ist die Molekülorbitaltheorie eine auf quantenmechanischen Grundlagen beruhende Theorie. Bei diesem Ansatz werden jedoch die Atome nicht getrennt betrachtet, sondern es wird zunächst aus den einzelnen Atomorbitalen ein Molekülorbital gebildet, in das die Elektronen gemäß der Hundschen Regel und dem Pauli-Prinzip eingeordnet werden.
Molekülorbitale werden nach der LCAO-Methode durch Linearkombination der beteiligten Atomorbitale gebildet. Durch die Linearkombination werden immer zwei Molekülorbitale, ein bindendes und ein antibindendes, gebildet, die der Addition beziehungsweise Subtraktion der Wellenfunktionen der beiden Atomorbitalen entsprechen.
- Ψb = ΨA + ΨB
- Bildung des bindenden Orbitals
- Ψa = ΨA − ΨB
- Bildung des antibindenden Orbitals
Wellenfunktion eines bindenden s-Orbitals
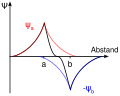
Wellenfunktion eines antibindenden s-Orbitals
Im bindenden Molekülorbital ist das Überlappungsintegral positiv und dadurch die Elektronendichte zwischen den Atomen erhöht. Durch die erhöhte Elektronendichte sind die Kerne voneinander besser abgeschirmt, was energetisch günstig ist. Es bildet sich daher eine Bindung aus. Im antibindenden ist das Überlappungsintegral dagegen negativ, es kommt zu einer Knotenebene und einer geringeren Elektronendichte zwischen den Kernen. Da dies energetisch ungünstig ist, kann sich keine Bindung ausbilden.
Werden in einem Molekülorbital die bindenden und antibindenden Orbitale mit Elektronen besetzt, lässt sich die Bindungsordnung bestimmen. Dazu wird die Anzahl an Elektronen in den antibindenden von der in den bindendene abgezogen. Ist die theoretische Bindungsordnung, wie beim hypothetischen He2-Molekül, null, ist das Molekül instabil.
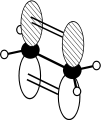
HOMO (bindend) der π-Bindung bei Ethen
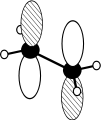
LUMO (antibindend) der π-Bindung bei Ethen
Kristallfeld- und Ligandenfeldtheorie
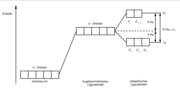 Aufspaltung im oktaedrischen Ligandenfeld
Aufspaltung im oktaedrischen Ligandenfeld- Hauptartikel: Kristallfeld- und Ligandenfeldtheorie
Die Kristallfeldtheorie und ihre Erweiterung, die Ligandenfeldtheorie, versuchen die Bindungssituation in Komplexen, also Verbindungen mit einem Metallzentrum, in der Regel einem Übergangsmetall, und darumliegenden Liganden, Nichtmetallatomen oder kleinen Molekülen wie etwa Chlorid oder Wasser zu erklären.
Im Gegensatz zu Molekülen sind bei Komplexen die d-Orbitale des Metallatoms entscheidend an der Bindung beteiligt. Diese werden in ihrer Energie durch elektrostatische Wechselwirkungen mit den Liganden beeinflusst. Je nach Geometrie des Komplexes wird die Energie der einzelnen Orbitale unterschiedlich stark erhöht. Daher kommt es zur Aufhebung der Entartung der verschiedenen Orbitale und zu einer für jede Komplexgeometrie typischen Aufspaltung der Orbitalenergien. Die Stärke der Aufspaltung hängt von der Art des Zentralions, seiner Oxidationsstufe, der Geometrie des Komplexes und Art der Liganden ab. Deren unterschiedliche Fähigkeit zur Energieaufspaltung ist in der spektrochemischen Reihe festgelegt.
Mit der Kristallfeldtheorie lassen sind viele Eigenschaften von Komplexen gut erklären. So lassen sich die Farbe, die magnetischen Eigenschaften und die Stabilität von Komplexen damit vorhersagen. Die Theorie ist allerdings begrenzt, so lässt sich etwa die besondere Stärke von Kohlenstoffmonoxid als Ligand und der nephelauxetische Effekt nicht über die Kristallfeldtheorie erklären. Genauere Ergebnisse liefert dafür die Molekülorbitaltheorie, bei der nicht nur die d-Orbitale des Zentralatoms, sondern alle beteiligten Orbitale in die Berechnungen mit einbezogen werden.[4]
Bindigkeit
Die Bindigkeit, also die Anzahl an Bindungen, die ein Atom eingehen kann, wird von den Orbitalen bestimmt. Es ist gemäß der Edelgasregel günstig, voll-, halbbesetzte oder leere Orbitale zu bilden. Daher nimmt ein Atom in der Regel so viele Elektronen von benachbarten Atomen auf und bildet Bindungen aus, bis es die Edelgaskonfiguration erreicht hat. Da die maximale Anzahl an Valenzelektronen, die ein Atom der 2. Periode erreichen kann, acht beträgt, wird häufig von der Oktettregel gesprochen. So besitzt beispielsweise Sauerstoff selbst sechs Valenzelektronen und kann noch zwei weitere aufnehmen. Dementsprechend bildet er auch in der Regel zwei Bindungen zu anderen Atomen aus, das stabile Oxid-Ion ist durch die Aufnahme zweier zusätzlicher Elektronen zweifach negativ geladen.
Metallische Bindung
- Hauptartikel: Metallische Bindung
Bei der Metallischen Bindung liegen im Gegensatz zur ionischen oder kovalenten Bindung frei bewegliche Elektronen vor, die nicht an ein bestimmtes Atom gebunden sind. Ein einfaches Modell ist das des Elektronengases, bei dem die Valenzelektronen ein negativ geladenes „Gas“ bilden, dass die positiv geladenen „Atomrümpfe“ vollständig umgibt und für den Ladungsausgleich sorgt.
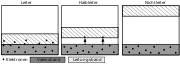 Bändermodell für Leiter, Halbleiter und Nichtleiter
Bändermodell für Leiter, Halbleiter und NichtleiterEin genaueres Modell ist das aus der Molekülorbitaltheorie ableitbare Bändermodell, das vorwiegend zur Beschreibung von Metallen verwendet wird, jedoch auch Eigenschaften von Salzen und Halbleitern erklären kann. Dabei werden aus den Valenzorbitalen bindende und antibindende Molekülorbitale gebildet. Auf Grund des Pauli-Prinzips besitzt jedes Atomorbital eine geringfügig andere Energie, so dass die Molekülorbitale im Metall breite Bänder bilden. Da in einem Metall nie alle Molekülorbitale vollständig besetzt sind, sind immer besetzte und unbesetzte Bänder im Metall vorhanden. Je nach Lage der Energieniveaus können die besetzten und unbesetzten Bänder einen weiten oder geringen Abstand haben oder auch, wie bei Metallen, überlappen. Die Überlappung der Bänder bei Metallen erklärt die gute Leitfähigkeit dieser, da für die Übertragung von Elektronen vom besetzten Valenzband in das unbesetzte Leitungsband keine Energie nötig ist. Bei Halbleitern, bei denen die Bänder einen geringen Abstand haben, kann dieser durch Energiezufuhr, beispielsweise durch Erhitzen, überwunden werden. Dies bedingt die temperaturabhängige Leitfähigkeit von Halbleitern. Salze dagegen besitzen einen zu großen Bandabstand, als dass er durch Wärmezufuhr überwindbar wäre und sind dementsprechend nicht leitend.
Schwache Bindungen
Schwache Bindungen werden manchmal auch zu den Bindungen gezählt, sind jedoch keine eigentlichen Bindungen innerhalb von Molekülen, sondern Wechselwirkungen, die zwischen verschiedenen Molekülen wirken und schon mit geringer Energiezufuhr gebrochen werden können. In der Regel reicht die Bewegungsenergie der Atome und Moleküle aus, um eine solche Bindung schon nach sehr kurzer Zeit wieder zu brechen. Die stärkste der schwachen Bindungen ist die Wasserstoffbrückenbindung. Diese bildet sich zwischen an elektronegative Atome gebundenen Wasserstoffatomen und freien Elektronenpaaren anderer elektronegativer Atome aus. Die bekannteste Verbindung, die Wasserstoffbrücken ausbildet, ist Wasser, aber auch Ammoniak, Fluorwasserstoff und andere Verbindungen, die N-H-, O-H- oder F-H-Bindungen besitzen, sind dazu in der Lage. Auf Grund ihrer Stärke beeinflusst die Wasserstoffbrückenbindung die Eigenschaften eines Moleküls stark, unter anderem ist sie für den ungewöhnlich hohen Schmelzpunkt des Wassers verantwortlich.
Deutlich schwächer sind die Van-der Waals-Wechselwirkungen, unter denen verschiedene Phänomene, etwa die London-Kräfte und Dipol-Wechselwirkungen zusammengefasst werden. Diese Kräfte wirken zwischen ungeladenen Molekülen und bewirken so, dass auch unpolare Substanzen als Flüssigkeit oder Feststoff vorliegen können.
Messung von Bindungseigenschaften
Eigenschaften einer chemischen Bindung können mit verschiedenen Messverfahren untersucht werden, die sich je nach Art der Verbindung und dem Aggregatzustand unterscheiden. Die Bindungslänge wird in Feststoffen mit Hilfe der Röntgenbeugung bestimmt. Dabei werden über die Elektronendichteverteilung die Positionen der Atome in einem Kristall bestimmt. Aus diesen Positionen lässt sich einfach der Abstand und damit die Bindungslänge ermitteln. Bei kleinen gasförmigen Molekülen lässt sich die Rotationsspektroskopie zur Ermittlung der Bindungslänge nutzen.
Die exakte Bindungsenergie lässt sich experimentell nicht bestimmen. Nährungsweise wird sie aus den Dissoziationsenthalpien möglichst einfacher Moleküle ermittelt, für genaue theoretische Werte müssen zusätzliche Faktoren, wie die Nullpunktsenergie, Rotationsenergien oder die Volumenarbeit beachtet werden.[6] Ähnliches gilt auch für die Gitterenergie, die sich nur indirekt über den Born-Haber-Kreisprozess bestimmen lässt.
Einzelnachweise
- ↑ a b c William H. Brock: Viewegs Geschichte der Chemie, Vieweg, Braunschweig 1997, S. 292-319, ISBN 3-540-67033-5.
- ↑ a b Friedrich Hund: Frühgeschichte der quantenmechanischen Behandlung der chemischen Bindung. In: Angewandte Chemie 1977, 89, S. 89-94, doi:10.1002/ange.19770890206
- ↑ Peter W. Atkins, Julio de Paula: Physikalische Chemie, 4. Auflage, Wiley-VCH, Weinheim 2006, S. 1129, ISBN 978-3-527-31546-8.
- ↑ a b J. Huheey, 3. Auflage, S. 480.
- ↑ a b J. Huheey, 3. Auflage, S. 1164-67.
- ↑ J. Huheey, 3. Auflage, S. 1155-1159.
Literatur
- Gernot Frenking: Chemische Bindung. In: Römpp Chemie-Lexikon, Thieme Verlag, Stand 2006, online.
- James E. Huheey, Ellen A. Keiter, Richard L. Keiter: Anorganische Chemie, 3. Auflage, de Gruyter, Berlin 2003, ISBN 3-11-017903-2.
- Arnold F. Holleman, Nils Wiberg: Lehrbuch der Anorganischen Chemie, 102. Auflage, de Gruyter, Berlin 2007, ISBN 978-3-11-017770-1.
- Werner Kutzelnigg: Einführung in die Theoretische Chemie, Teil II: Die chemische Bindung, Wiley-VCH, Weinheim 2002, ISBN 3-527-30609-9.
- Joachim Reinhold: Quantentheorie der Moleküle, 3. Auflage, Teubner, Wiesbaden 2006, ISBN 3-83-510037-8.
- Linus Pauling: Die Natur der chemischen Bindung, 2.Nachdr. d. 3. Auflage, Wiley-VCH, Weinheim 1988, ISBN 978-3-527-25217-6.
Weblinks


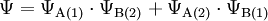







Wikimedia Foundation.