- Atypischer teratoider/rhabdoider Tumor
-
Klassifikation nach ICD-10 C72 Bösartige Neubildung des Gehirns
ICD-O 9508/3
WHO Grad IVICD-10 online (WHO-Version 2011)  Magnetresonanztomographie eines großen atypischen teratoiden/rhabdoiden Tumors bei einem Säugling.
Magnetresonanztomographie eines großen atypischen teratoiden/rhabdoiden Tumors bei einem Säugling.
Als atypischer teratoider/rhabdoider Tumor, häufig auch als ATRT oder AT/RT abgekürzt, wird ein seltener Hirntumor aus der Gruppe der embryonalen Tumoren bezeichnet. Der äußerst bösartige Tumor wird nach der WHO-Klassifikation der Tumoren des zentralen Nervensystems als Grad IV eingeteilt und tritt ganz überwiegend bei Kleinkindern auf.
Inhaltsverzeichnis
Epidemiologie
Atypische teratoide/rhabdoide Tumoren machen etwa 2% aller Hirntumoren bei Kindern aus, wobei ganz überwiegend Kinder in den ersten Lebensjahren betroffen sind.[1] Bei Erwachsenen ist das Auftreten nur in Einzelfällen beschrieben.[2]
Sporadische Fälle, bei denen kein Zusammenhang mit einer erblichen Erkrankung erkennbar ist, überwiegen. Eine familiäre Häufung tritt beim sogenannten Rhabdoid-Prädispositions-Syndrom auf.[3]
Symptomatik
Atypische teratoide/rhabdoide Tumoren können in der hinteren Schädelgrube lokalisiert sein, treten aber auch im Bereich der Großhirnhemisphären auf. Die klinische Symptomatik ist abhängig von der Tumorlage und dem Alter des Kindes und wird in der Regel durch das rasche Tumorwachstum geprägt. Schläfrigkeit, Übelkeit und Erbrechen sind häufige, aber unspezifische Symptome.
Neuropathologie
 Histologie eines ATRT mit zahlreichen rhabdoiden Tumorzellen. Hämatoxylin-Eosin gefärbtes Schnittpräparat.
Histologie eines ATRT mit zahlreichen rhabdoiden Tumorzellen. Hämatoxylin-Eosin gefärbtes Schnittpräparat.Namensgebend und für die feingeweblichen (histologischen) Eigenschaften typisch sind relativ große Tumorzellen, die randständige, hervorstechende Kernkörperchen (Nukleolen) und Einschlüsse in ihrem Zellkörper besitzen, sogenannte rhabdoide Tumorzellen. Sie erinnern in ihrem Zellbild an einen bösartigen Weichteiltumor, das Rhabdomyosarkom. Mitotische Aktivität, Zelldichte und Kernpolymorphie sind erhöht. Tumornekrosen sind häufig. Der Tumor wächst unstrukturiert oder in papillären Strukturen, was die Abgrenzung gegenüber Plexuskarzinomen schwierig machen kann. Andererseits kommen neben Abschnitten mit rhaboiden Tumorzellen häufig auch kleinzellige Anteile vor, deren Histologie dem anderer embryonaler Tumoren wie dem Medulloblastom und dem sPNET ähneln.
Pathogenese
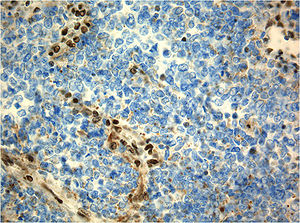 Immunhistochemie für INI1, das Produkt des bei ATRT typischerweise defekten SMARCB1-Gens. Beachte die fehlende Braun-Anfärbung der Zellkerne der Tumorzellen im Vergleich zu den kräftig braungefärbten Zellkernen der mitgetroffenen Blutgefäße (interne Positivkontrolle).
Immunhistochemie für INI1, das Produkt des bei ATRT typischerweise defekten SMARCB1-Gens. Beachte die fehlende Braun-Anfärbung der Zellkerne der Tumorzellen im Vergleich zu den kräftig braungefärbten Zellkernen der mitgetroffenen Blutgefäße (interne Positivkontrolle).Genetische Veränderungen die das SMARCB1-Gen auf Chromosom 22q11.2 betreffen und zu einer verminderten Expression dessen Genprodukts, des INI1-Proteins, führen, sind charakteristisch. Bei etwa einem Drittel der Patienten ist eine Keimbahnmutation des SMARCB1-Gens nachweisbar. Dieser Befund geht mit jüngerem Erkrankungsalter und einer noch ungünstigeren Prognose einher.[4]
Der immunhistochemische Nachweis einer fehlenden INI1-Proteinexpression läßt sich in der Mehrzahl der Fälle zur Abgrenzung gegenüber anderen Tumoren diagnostisch nutzen.[5][6] Da ATRT jedoch selten auch in Zusammenhang mit genetischen Veränderungen von SMARCA4 (BRG1), einem Gen, das für eine weitere Unterheit des SWI/SNF Chromatin-Remodeling-Complex kodiert, auftreten können,[7] macht der Nachweis einer erhaltenen INI1-Expression die Diagnose eines ATRT zwar unwahrscheinlicher, schließt sie jedoch keinesfalls aus.[8]
Behandlung und Prognose
Eine vollständige operative Entfernung kann häufig nicht erreicht werden. Im Rahmen der weiteren Behandlung, die in der Regel im Rahmen klinischer Studien erfolgt, schließt sich deswegen eine adjuvante Chemotherapie und (im Rahmen eines individuellen Behandlungskonzepts in Abhängigkeit vom Alter des Kindes) möglicherweise auch eine Bestrahlung an.
Auch nach operativer Entfernung und initial gutem Ansprechen auf die Chemotherapie ist der weitere Verlauf häufig von einem erneutem Tumorwachstum geprägt. Zwei Jahre nach Diagnosestellung waren im Rahmen einer klinischen Studie trotz intensiver therapeutischer Bemühungen 83% der betroffenen Kleinkinder gestorben. Kinder mit einem Alter von über drei Jahren haben eine etwas bessere Prognose, was möglicherweise durch die (aufgrund der Nebenwirkungen erst in dieser Altersgruppe durchführbaren) Bestrahlung bedingt ist.[9]
Durch eine intensive und aggressive Kombinationsbehandlung konnte im Rahmen einer an amerikanischen Kliniken durchgeführten klinischen Studie eine Zweijahresüberlebensrate von 70% erreicht werden.[10]
Literatur
- Rorke & Biegel: Atypical teratoid/rhabdoid tumour. In: Kleihues, Cavenee, eds. World Health Organization classification of tumors. Pathology and genetics of tumours of the nervous system. Lyon, IARC Press; 2000
Einzelnachweise
- ↑ Strother D:Atypical teratoid rhabdoid tumors of childhood: diagnosis, treatment and challenges. Expert Rev Anticancer Ther. 2005;5(5):907-15. PMID 16221059 (Übersichtsarbeit)
- ↑ Makuria et al.: Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol. 2008;88(3):321-30. PMID 18369529
- ↑ Sévenet et al.: Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999;65(5):1342-8. PMID 10521299
- ↑ Bruggers CS, Bleyl SB, Pysher T, et al.: Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. In: Pediatr Blood Cancer. September 2010. doi:10.1002/pbc.22757. PMID 20848638.
- ↑ Judkins et al.:INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. J Neuropathol Exp Neurol. 2005;64(5):391-7.PMID 15892296
- ↑ Haberler et al.:Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: Lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol. 2006;30(11):1462-8. PMID 17063089
- ↑ R. Schneppenheim, M. C. Frühwald, S. Gesk, M. Hasselblatt, A. Jeibmann, U. Kordes, M. Kreuz, I. Leuschner, J. I. Martin Subero, T. Obser, F. Oyen, I. Vater, R. Siebert: Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. In: Am. J. Hum. Genet. 86, 2010, S. 279–284, PMID 20137775 PMC 282019.
- ↑ Pfister SM, Korshunov A, Kool M, Hasselblatt M, Eberhart C, Taylor MD: Molecular diagnostics of CNS embryonal tumors. In: Acta Neuropathol.. 120, Nr. 5, November 2010, S. 553–66. doi:10.1007/s00401-010-0751-5. PMID 20882288.
- ↑ Tekautz et al.: Atypical teratoid/rhabdoid tumors (ATRT): improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol. 2005;23(7):1491-9. PMID 15735125
- ↑ Chi et al.: Intensive Multimodality Treatment for Children With Newly Diagnosed CNS Atypical Teratoid Rhabdoid Tumor. J Clin Oncol. 2009;27(3):385-9. PMID 19064966
Weblinks
- EURHAB Europäisches Rhabdoidtumor-Register

Bitte den Hinweis zu Gesundheitsthemen beachten! Kategorien:- Krebserkrankung
- Tumor des zentralen Nervensystems
- Kinderonkologie



Wikimedia Foundation.