- Th1
-
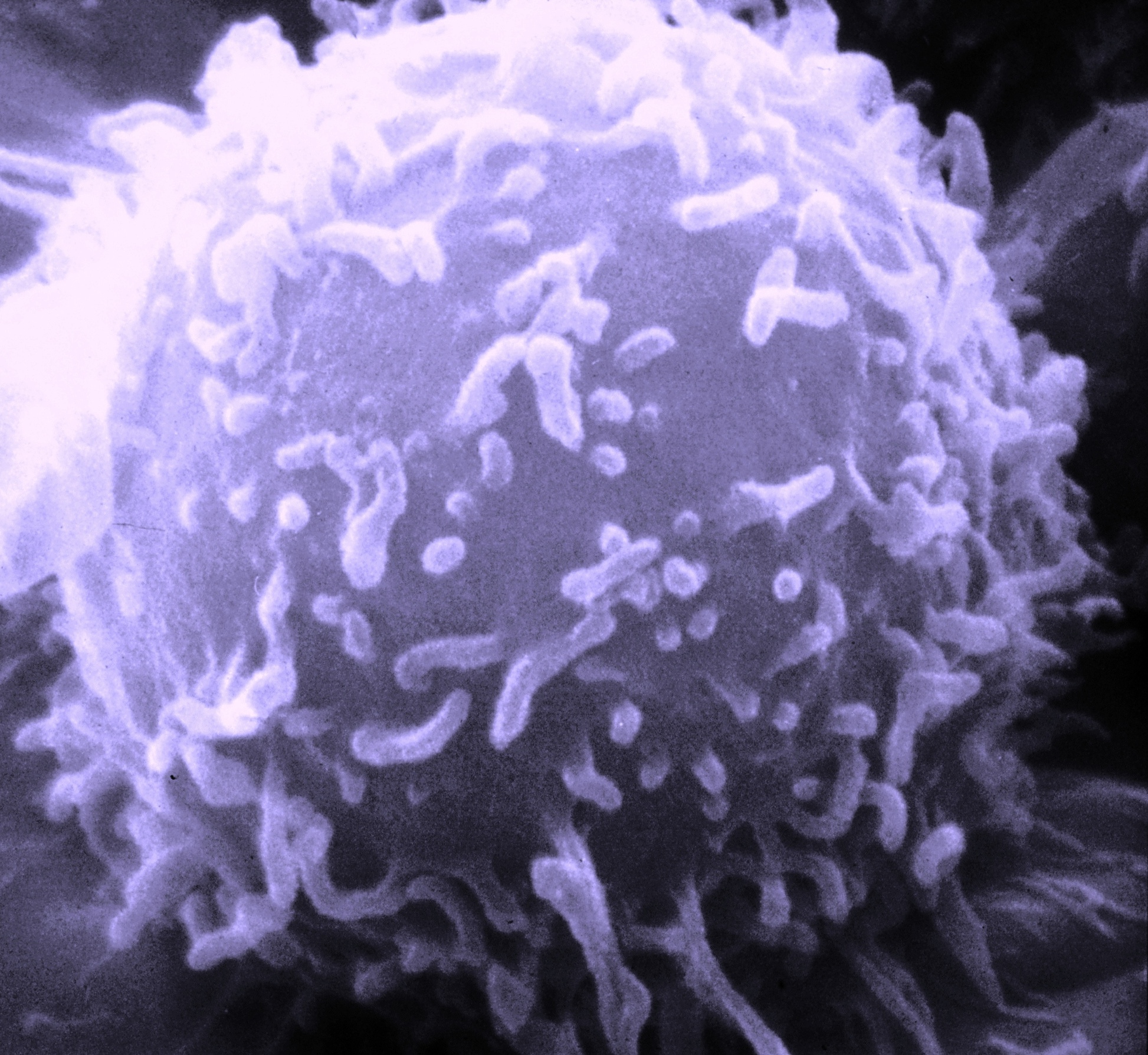
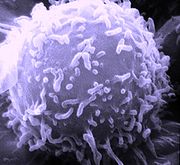 REM-Aufnahme eines Lymphozyten
REM-Aufnahme eines LymphozytenT-Lymphozyten oder kurz T-Zellen (T für Thymus-abhängig) sind eine für die Immunabwehr wichtige Gruppe von Blutzellen. Sie gehören zu den weißen Blutkörperchen (Leukozyten) und sind neben den B-Lymphozyten an der erworbenen Immunantwort beteiligt. Die Vorläufer der T-Zellen stammen, wie alle Blutzellen, aus dem Knochenmark. Von dort wandern sie in den Thymus, wo sie fast vollständig ausreifen. Es werden mehrere Unterarten der T-Zellen unterschieden, die unterschiedliche Funktionen im Immunsystem wahrnehmen.
T-Lymphozyten können mit Hilfe spezieller Moleküle auf ihrer Oberfläche, den T-Zell-Rezeptoren (TCR), so genannte Antigene erkennen, bei denen es sich in der Regel um körperfremde Stoffe handelt. Anders als die von B-Lymphozyten produzierten Antikörper, erkennen T-Lymphozyten aber keine ungebundenen Antigene, sondern nur solche, die ihnen mittels Proteinkomplexen (MHC) auf der Oberfläche Antigenpräsentierender Zellen vorgezeigt werden.
Funktion der T-Zellen
T-Zellen haben die Aufgabe, die Zellen des Körpers zu überwachen und zu reagieren, wenn in diesen Zellen unnatürliche Veränderungen auftreten. Diese Veränderungen können durch ein Pathogen, welches die Zelle befällt, oder durch genetische Veränderungen (Mutationen) hervorgerufen werden. Zu diesem Zweck wandern die T-Zellen durch Blutgefäße und Gewebe und tasten die Zelloberflächen der Zellen auf das Vorhandensein von MHC-I- oder MHC-II-Molekülen ab. Erkennt die T-Zelle das Signal eines MHC-Moleküls, so wird diese nur dann aktiv, wenn deren TCR nach dem Schlüssel/Schloss Prinzip zu dem präsentierten Antigen passt und zusätzlich von der APC eine Co-Stimulanz (etwa das Oberflächenprotein B7) präsentiert wird. Nur wenn alle Bedingungen erfüllt sind, geht die T-Zelle in den aktivierten Zustand über, und es wird ein Signal zum eigenen Zellkern gesendet. Dort wird entschieden, wie diese auf das Pathogen reagiert. Einige T-Zellen – T-Killerzellen mit CD8-Rezeptor – können die Pathogene eigenständig bekämpfen, während andere T-Lymphozyten – T-Helferzellen mit CD4-Rezeptor – an ihre Umgebung verschiedene lösliche Botenstoffe (Zytokine) abgeben. Damit aktivieren sie zusätzliche Immunzellen, die ihnen helfen, die Pathogene zu eliminieren.
Anschaulich aber auch vereinfacht lässt sich sagen: Körperzellen präsentieren eigene Stoffwechselprodukte auf MHC-I Molekülen. Outet sich eine Zelle hierdurch als defekt, soll die defekte Zelle durch andockende T-Zellen ausgeschaltet werden. Immunzellen präsentieren zusätzlich auf MHC-II Molekülen die Überbleibsel bekämpfter Feinde. Diese ja erfolgreichen Immunzellen sollen nun von andockenden T-Zellen nicht etwa ausgeschaltet, sondern im Gegenteil angespornt werden. Hieraus ergibt sich, dass T-Zellen bezüglich ihrer Funktion heterogen sein müssen. CD8-T-Zellen, die über MHC-I defekte Körperzellen erkennen, deaktivieren diese. CD4-T-Zellen, die über MHC-II erfolgreiche Immunzellen erkennen, aktivieren diese.
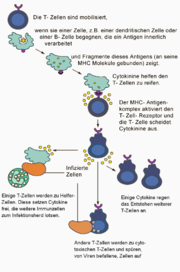 Aktivierungsablauf der reifen T-Lymphozyten
Aktivierungsablauf der reifen T-LymphozytenAls primäres lymphatisches Organ erlaubt der Thymus die gezielte Ausbildung von T-Zellen mit unterschiedlichen Effektorfunktionen. Zytotoxische T-Zellen erkennen und töten Zielzellen, welche Fremd-Antigene an ihrer Zelloberfläche besitzen; T-Helferzellen stimulieren die humorale Antwort gegen komplexe Antigene, beziehungsweise induzieren über die Stimulation von B-Zellen eine zelluläre Immunabwehr; regulatorische T-Zellen sorgen in einer antigenspezifischen Weise dafür, die T-Zell-Toleranz gegenüber Selbst-Antigenen aufrecht zu erhalten. Die Aufgabe, über die große Vielfalt unterschiedlicher TCR, eine größtmögliche Anzahl von Fremdantigenen erkennen zu können, wird durch T-Lymphozyten wahrgenommen. Diese T-Lymphozyten sind somit für die Regulation der humoralen Immunantwort, die zellvermittelte Zytotoxizität und die Überempfindlichkeitsreaktion verantwortlich.
Naive T-Zellen müssen zuerst aktiviert werden, bevor sie ihre Effektorfunktionen wahrnehmen. Die über den Antigenrezeptor und die Korezeptoren erhaltenen Signale kontrollieren die molekularen Vorgänge, welche schließlich zu Zellwachstum und Differenzierung der T-Zellen führen. Die Vermehrung der Zellen sichert, dass genügend T-Zellen zur Abwehr bereitstehen. Der genaue Charakter der T-Zell-vermittelten Leistung des Immunsystems ist vor allem auch von der Natur des stimulierenden Antigens, der Art der antigenpräsentierenden Zelle und weiterer intrinsischer Faktoren abhängig, die während der primären Immunantwort die Differenzierung der T-Zelle mit beeinflussen.
Durch die thymischen Selektionsvorgänge und die damit verbundene Expression der Oberflächenantigene CD4 und CD8 können αβ-Antigenrezeptor-positive T-Lymphozyten phänotypisch in unterschiedliche Subpopulationen eingeteilt werden. CD4+CD8--T-Lymphozyten gelten funktionell in der Regel als Helferzellen und besitzen einen Rezeptor, welcher Antigene im Kontext von MHC-Klasse-II-Molekülen erkennt. Zytotoxische T-Zellen sind im Gegensatz hierzu allgemeinen CD4-CD8+-T-Lymphozyten und besitzen einen MHC-I erkennenden T-Zell-Rezeptor.
Diese Unterteilung ist jedoch sehr vereinfachend und gelegentlich sogar falsch, denn sie spiegelt einzig die häufigste Korrelation zwischen T-Zell-Phänotyp und Funktion wider, zumal auch CD4+ zytotoxische T-Zellen und CD8+-T-Helferzellen bekannt sind. CD4+-T-Zellen finden sich vornehmlich im peripheren Blut und in jenen Abschnitten des lymphatischen Gewebes, welche einen großen Durchfluss von T-Lymphozyten aufweisen, wie etwa parafollikulären Regionen von Lymphknoten, Milz und Tonsillen. Im Gegensatz hierzu sind die CD8+-T-Zellen typischerweise im Knochenmark und im Bereich der Mukosa des Darm-Magen-Traktes, der Atemwege und der Harnwege lokalisiert. Naive T-Zellen zirkulieren kontinuierlich zwischen dem Blut und den lymphatischen Geweben, wobei sie die Gefäße im Bereich der hochendothelialen Venole (HEV) verlassen, aber später über die Lymphe und den Ductus thoracicus wieder ins Blut zurückkehren. Dieses Migrationsverhalten naiver T-Zellen wird durch die Expression von Zelladhäsionsmolekülen und Rezeptoren für Chemokine ermöglicht.
Die T-Lymphozyten haben eine geringe amöboide Beweglichkeit, sie können aus den Kapillaren in die Gewebe auswandern und umgekehrt aus den lymphatischen Organen in die Blut- und Lymphbahnen eindringen.
Immunbiologische Funktionen, welche den γδ-T-Zellen zugeschrieben werden, sind vergleichbar mit jenen der αβ-T-Zellen und beinhalten neben der Sekretion von Zytokinen auch die Immunregulation von T- und B-Lymphozyten und die Zytotoxizität. Die von den αβ-T-Zellen bekannte funktionelle Dichotomie in Typ1- und Typ2-polarisierten Zellen gilt auch für die Population der γδ-T-Zellen. γδ-T-Zellen produzieren innerhalb von 1-2 Tagen nach einer Infektion wesentliche Mengen an Gamma-Interferon und anderen proinflammatorische Zytokinen und helfen so nicht nur bei der Aktivierung von NK-Zellen und Makrophagen, sondern unterstützen auch die Stimulation von αβ-T-Zellen.
Spezielle Funktionen der T-Lymphozyten
Die Erkenntnis, dass Immunzellen in der Lage sind, Substanzen freizusetzen (sezernieren), die den Knochenstoffwechsel in nachhaltiger Weise beeinflussen können, hat der Verbindung zwischen Immunsystem, Sexualhormonen und Knochenstoffwechsel eine neue Bedeutung verliehen. Die Stimulation der TNF-α und Osteoprotegerin (OPGL)-Produktion von T-Lymphozyten stellt möglicherweise einen wichtigen Faktor in der Pathogenese des durch Östrogenmangel hervorgerufenen Knochenverlustes bei der Maus dar. So konnte gezeigt werden, dass athymische, T-Zell-defiziente Mäuse keinen Knochenverlust nach der operativen Entnahme der Eierstöcke (Ovariektomie) zeigen. Außerdem besitzen athymische Nacktmäuse, sowie athymische Nacktratten generell einen niedrigeren Knochenturnover (Knochenstoffwechsel). Durch die Erkenntnis, dass aktivierte T-Lymphozyten OPLG sezernieren und so eine Stimulation der osteoklastären Knochenresorption induzieren können, geht man heute davon aus, dass dieser Mechanismus entscheidend an der Pathogenese von Knochen- und Gelenk-Erkrankungen beteiligt ist.[1][2]
Aufbau und Unterscheidung von verwandten Zelltypen
Die T-Zellen gehören zu den Lymphozyten und sind kugelige Zellen, die einen etwa gleich großen Durchmesser haben wie Erythrozyten (beim Menschen etwa 7,5 µm). Der Zellkern, in welchem der Nucleolus schwer zu sehen ist, zeigt eine relativ dichte, schollige Chromatinstruktur und bekommt dadurch eine kräftige Färbung. Er ist mehr oder weniger kugelig, häufig an einer Stelle etwas eingedellt, aber niemals gelappt. Das an freien Ribosomen reiche Zytoplasma bildet um den Kern einen hellen, schmalen Saum, der lichtmikroskopisch oft kaum sichtbar ist. T-Zellen besitzen kleine Lysosomen (azurophile Granula). Der Golgi-Apparat der Zellen ist kleiner als bei den Retikulumzellen.
Morphologisch und elektronenmikroskopisch unterscheiden sich T-und B-Lymphozyten nicht. Funktionell gibt es aber Unterschiede: Im Gegensatz zu den B-Lymphozyten erkennen T-Zellen Antigene ausschließlich in Form von Oligopeptiden, die ihnen gemeinsam im Komplex mit körpereigenen Histokompatibilitäts-Antigen (MHC) präsentiert werden; eine Einschränkung, welche als MHC-Restriktion bezeichnet wird.
Die Strukturen an der Oberfläche von T-Lymphozyten, welche diesen Komplex aus körperfremdem (Peptid-Antigen) und körpereigenen (MHC-Molekül) Anteil spezifisch erkennen, werden als T-Zell-Antigenrezeptoren (TCR) bezeichnet. Eine Bestimmung der verschiedenen Lymphozytentypen (NK-Zellen, B-Lymphozyten, T-Lymphozyten) kann auf Grund von Markerproteinen (CD-Antigene) auf ihrer Zelloberfläche vorgenommen werden, diese können für die Untersuchung durch monoklonale Antikörper markiert werden.
Die anatomische Verteilung der T-Lymphozyten
Die Mehrzahl der T-Lymphozyten in der Peripherie des Blutkreislaufs gehört zu den αβ-T-Zellen (95–98 %), dagegen nehmen γδ-T-Zellen nur einem kleinen Bestandteil (1–5 %) an den Lymphozyten des peripheren Blutes und der konventionellen lymphatischen Organe wie Milz und Lymphknoten ein. Im Gegensatz dazu nimmt der γδ-T-Zelltyp einen hohen Anteil (bis zu 50 %) in epithelialen Geweben wie der Haut, der Darmschleimhaut oder den Geschlechtsorganen ein. Aus dieser Verteilung wird vermutet, dass γδ-T-Zellen zur immunologischen Überwachung der Körperoberflächen dienen.
Die T-Zell-Entwicklung
Die zentrale (thymische) T-Zell-Entwicklung
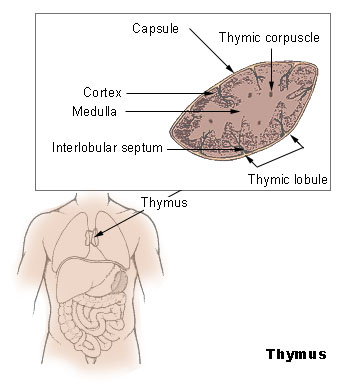
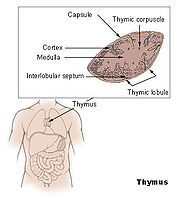 Anatomie des Thymus
Anatomie des ThymusDer Thymus ist das primäre lymphatische Organ, in dem die Entwicklung von Vorläuferzellen zu funktionellen Antigen-spezifischen T-Lymphozyten erfolgt. Die als Thymozyten bezeichneten lymphatischen Vorläuferzellen durchlaufen unterschiedliche Entwicklungsstadien, die durch das Exprimieren von Oberflächenmarkern und spezifischen molekularen Eigenschaften gut definiert sind. Die für die Ausreifung notwendigen Stimuli werden vornehmlich durch hämatopoetische und epitheliale Stromazellen bereitgestellt. Die Interaktionen zwischen Stromazellen und sich entwickelnden Thymozyten ist auch für die komplexen Vorgänge der positiven und negativen Selektion von Thymozyten verantwortlich.
Normalerweise wird die Thymusanlage ab der sechsten Schwangerschaftswoche durch T-Lymphozyten-Vorläuferzellen der Leber und ab der 22. Schwangerschaftswoche durch die Zellpopulation aus dem Knochenmark besiedelt. Sobald Blutgefäße in den Thymus einsprossen, erfolgt über sie die Einwanderung von T-Vorläuferzellen. Auf diesen frühesten T-Lymphozyten-Vorläuferzellen können die Chemokin-Rezeptoren CXCR4 und CCR9 nachgewiesen werden. Der ausgebildete Thymus besteht aus mehreren, von Trabekeln (Scheidewänden aus Bindegewebe) begrenzten Läppchen, die ihrerseits – nach morphologischen und zellphänotypischen Kriterien – in einen äußeren Rindenbereich (kortikal) und einen zentralen Mark-Bereich (medullär) unterteilt werden. Die hämatopoetischen Vorläuferzellen gelangen über Transmigration durch die Endothelzellen der Blutgefäße in den unmittelbaren Bereich der Übergangszone zwischen Rinde und Mark, wo sie als pluripotente unreife lymphoide Zellen mit ihrer linienspezifischen Ausreifung beginnen. Im weiteren Verlauf des Reifungsprozesses wandern diese Zellen über die Rinde in das Mark des Thymus. Parallel zu dieser Wanderung erfolgt die Ausdifferenzierung der Thymozyten, bis die reifen T-Zellen aus dem Thymus schließlich in die Peripherie auswandern (Emigration).
Reifung der Thymozyten der αβ-T-Zell-Linie
Über die differenzielle Oberflächenexpression der Korezeptoren CD4 und CD8 werden vier wesentliche Untergruppen von Thymozyten unterschieden, die alle wichtigen Reifungsstadien der Entwicklung innerhalb des Thymus der αβ-T-Zell-Linie definieren. Das früheste intrathymische Entwicklungsstadium ist einerseits durch das gleichzeitige Fehlen der CD4- und CD8-Expression charakterisiert, weshalb diese Zellen auch als doppelt-negative (DN) Thymozyten bezeichnet werden, und andererseits durch den Mangel eines vollständigen T-Zell-Antigenrezeptors gekennzeichnet. Aus den DN-Zellen (etwa 3–5 % aller Thymozyten) entwickeln sich die so genannten doppelt-positiven (DP) CD4+-CD8+-Thymozyten, welche die zahlenmäßig größte Zellpopulation (etwa 85 %) darstellen. Aus den DP-Thymozyten entwickeln sich schließlich die reifen, so genannten einfach-positiven (single positive SP) CD4+-CD8-- (etwa 8 %) beziehungsweise CD4--CD8+-T-Zellen (etwa 4 %). Jede dieser vier Subpopulationen kann noch durch zusätzliche phänotypische Marker weiter unterteilt werden.
Positive thymische Selektion
Durch den Prozess der positiven thymischen Selektion wird erreicht, dass jeder T-Zell-Antigenrezeptor die körpereigenen Haupthistokompatibilitätskomplex-Moleküle (MHC) und die in der Antigenbindungsgrube enthaltenen Selbst-Antigene als Komplex mit adäquater Affinität erkennen kann. Die für die positive Selektion benötigten Peptid/MHC-Komplexe werden durch thymische Epithelzellen bereitgestellt. Die hierzu wesentliche Avidität zwischen Thymozyten und Epithelzellen wird durch die Affinität des Antigenrezeptors zu seinen spezifischen Peptid/MHC-Liganden an der Epithelzelloberfläche und durch die Konzentration der Korezeptoren während der Zellbindung bestimmt. Nur wenn bei der Antigenerkennung die Interaktion des T-Zell-Antigenrezeptors über eine weder zu starke noch zu geringe Avidität erfolgt, werden die Überlebenssignale zur weiteren Entwicklung der DP-Thymozyten bereitgestellt. Obwohl Selbst-Peptide im Wesentlichen die stabile Oberflächenexpression und die konkrete molekulare Konformation der MHC-Moleküle beeinflussen, ist interessanterweise ihre direkte Erkennung durch den T-Zell-Antigenrezeptor für die Vorgänge der positiven Selektion von geringer Bedeutung. In der Tat können bereits einzelne wenige Peptide eine enorme Vielzahl von Thymozyten mit unterschiedlichen T-Zell-Antigenrezeptoren-Spezifitäten positiv selektionieren. Nach der positiven Selektion ist eine erneute Rekombination der α-Kette des Antigenrezeptors nicht mehr möglich. Durch diesen Vorgang wird es sichergestellt, dass T-Zellen mit einem positiv selektionierten Rezeptor ihre Spezifität nicht mehr ändern können. Thymozyten, welche den Komplex aus Selbst-Peptiden/MHC-Molekülen über ihren T-Zell-Antigenrezeptor nicht oder nur mit ungenügender Affinität erkennen können, generieren keine der für das weitere Überleben notwendigen Signale und sterben in der Folge durch den programmierten Zelltod.
Nach erfolgter positiver Selektion ist das Repertoire der T-Zell-Antigenrezeptoren nun so weit eingeschränkt, dass alle reiferen Thymozyten über einen Rezeptor verfügen, welcher auf den eigenen MHC-Komplex ausgerichtet ist. Die positive Selektion stellt es auch sicher, dass CD8+- und CD4+-Zellen am Ende ihrer Reifung eine Antigenpräsentation durch MHC-Klasse-I/II-Molekülen erkennen können.
Negative thymische Selektion
In einem zweiten, als negative thymische Selektion bezeichneten Prozess werden nun jene αβ-T-Zell-Antigenrezeptor-positiven Thymozyten ausgeschieden, welche einen Antigenrezeptor exprimieren, dessen Spezifität gegen körpereigene Proteine gerichtet ist. Dabei muss der T-Zell-Antigenrezeptor nicht notwendigerweise eine hohe Affinität für ihn spezifischen Antigen/MHC-Komplex aufweisen, solange diese Moleküle in hoher Konzentration an der Zelloberfläche exprimiert werden. Die als hämatopoetische Stromazellen im Thymus wirkenden dendritischen Zellen und Makrophagen sind typischerweise für die negative Selektion verantwortlich, doch können auch medulläre Epithelzellen die Eliminierung autoreaktiver T-Zellen vornehmen. Jene DP-Thymozyten, welche nun den Komplex aus Selbst-Peptid/MHC mit zu großer Avidität erkennen, werden im Bereich der Thymusrinde, an der Mark-Rinden-Übergangszone und des Marks durch Zelltod eliminiert. Neben der beschriebenen Deletion von autoreaktiven T-Zellen durch den Prozess der Apoptose, können reifende T-Zellen mit einem gegen Selbst-Antigene gerichteten Antigenrezeptor auch über den Vorgang der Anergie daran gehindert werden, als reife Zellen autoimmunpathologische Schäden in der Peripherie zu verursachen.
Am Ende der Entwicklung entstehen solche T-Lymphozyten die MHC-gebundene Liganden mit hoher Affinität, aber keine körpereigene Proteine erkennen können.
Die Entwicklung der γδ-T-Zellen
Während der Embryogenese werden γδ-T-Zellen vor der Bildung der αβ-Subpopulation gebildet. Diese Zellen migrieren in zeitlich bestimmten Schüben zuerst zu den inneren und äußeren Körperoberflächen (Haut, Schleimhaut) und können erst später auch im klassischen, sekundären lymphatischen Gewebe von Milz und Lymphknoten nachgewiesen werden. γδ-T-Zellen werden sowohl im Thymus als auch extrathymisch gebildet. Im Thymus können Vorläuferzellen identifiziert werden, welche noch das Potential besitzen, sich sowohl zu αβ- als auch zu γδ-T-Zellen entwickeln zu können.
Der T-Zell-Antigenrezeptor (TCR)
Hauptartikel: T-Zell-Rezeptor
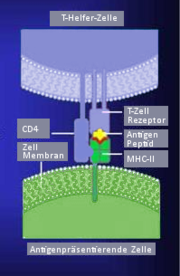 Von der antigenpräsentierenden Zelle wird das Antigen in Verbindung mit MHC-II dem T-Zell-Antigenrezeptor vorgezeigt
Von der antigenpräsentierenden Zelle wird das Antigen in Verbindung mit MHC-II dem T-Zell-Antigenrezeptor vorgezeigtGenau wie der Antigenrezeptor der B-Lymphozyten gehört der TCR zur Immunglobulin-Superfamilie, sein spezifisches Antigen kann er jedoch nur im Komplex mit MHC-Proteinen erkennen. Die Identifizierung des TCR gelang mit Hilfe von Antikörpern, die an bestimmte T-Zell-Klone binden. Der TCR besteht aus zwei Ketten, meist α und β, die über Disulfidbrücken verbunden sind. Die α- und die β-Kette haben, je nach Herkunft, eine molare Masse von 43-49 beziehungsweise 38-44 Kilodalton. Außer dem üblichen αβ- Heterodimer können T-Zellen noch ein γδ-Heterodimer bilden. Die γ-Kette ist 55-60 Kilodalton und die δ-Kette ist ca. 40 Kilodalton groß. Die gleichzeitige Expression von γδ- und αβ- Heterodimer auf der gleichen Zelle ist aufgrund der Organisation der α- und δ-Ketten-Gene nicht möglich. Die Gene für die α- und δ-Kette liegen beim Menschen ineinander geschachtelt am gleichen Genort auf dem Chromosom 14q11-12. Das γ-Ketten-Gen liegt auf Chromosom 7p15 und das β-Ketten-Gen auf Chromosom 7q32-35.
Kristallographische Untersuchungen zur dreidimensionalen Struktur haben Ähnlichkeiten zwischen dem TCR und den Antikörpern aufgezeigt. So ist die für die MHC/Antigen-Bindung wichtige V-Region des TCR vergleichbar mit der entsprechenden V-Domäne der Antikörper. An der Kontaktstelle zwischen TCR und MHC/Antigen-Komplex befinden sich hypervariable Molekülabschnitte, welche die Antigenspezifität des TCR bestimmen. Strukturell bilden diese Abschnitte exponierte Schleifen, welche als komplementaritätsbestimmende Regionen (Complementarity determining regions, CDR) bezeichnet werden. Der gemeinsam von T-Zell-Antigenrezeptor und MHC/Peptid gebildete Komplex besitzt eine Länge von 15 nm und ist somit im Vergleich zu anderen membranständigen Molekülen eher klein.
Die Interaktion des TCR mit dem MHC-Komplex (und damit auch die Aktivierung der T-Lymphozyten) wird im Wesentlichen durch drei Faktoren bestimmt: die TCR-Dichte auf der Oberfläche von T-Zellen, die Dichte spezifischer MHC/Antigen-Komplexe auf der Oberfläche von antigenpräsentierenden Zellen, und die Affinität des TCR für den MHC/Antigen-Komplex.
Der TCR liegt als Komplex mit dem CD3-Rezeptor vor. Während der TCR den MHC/Antigen-Komplex erkennt, ist der CD3-Rezeptor mit seinen sechs Untereinheiten für die Weiterleitung von aktivierenden Signalen in das Zellinnere verantwortlich. Der CD3-Rezeptor bindet sowohl TCRαβ- als auch TCRγδ-Ketten und findet sich auf allen peripheren T-Zellen und einem Teil der Thymozyten. Fehlt der CD3-Komplex, dann werden auch keine TCR-Ketten auf der Zelloberfläche exprimiert.
Unterarten der T-Lymphozyten
Die T-Lymphozyten werden anhand des Aufbaus ihres T-Zell-Antigenrezeptors in zwei Subpopulationen unterteilt. Der größere Anteil der T-Lymphozyten sind die αβ-Antigenrezeptor-positiven T-Lymphozyten (etwa 90 %), während γδ-Antigenrezeptor-positive T-Lymphozyten nur etwa 5–10 % der Gesamtpopulation der T-Zellen ausmachen [3].
αβ-Antigenrezeptor-positive T-Lymphozyten
T-Helferzellen
- → Hauptartikel: T-Helferzelle
T-Zellen mit einer Helferfunktion werden aufgrund der von ihnen sezernierten Zytokine hauptsächlich in zwei Subpopulationen unterteilt, die jeweils unterschiedliche Funktionen wahrnehmen. Die eine Subpopulation ist an der Ausbildung einer zellvermittelten Immunantwort beteiligt, während die andere Subpopulation die Gestaltung der humoralen Immunantwort mit bestimmt. Diese funktionelle Dichotomie in die so genannten Typ1- beziehungsweise Typ2-T-Zellen ist exemplarisch für CD4+-Lymphozyten beschrieben worden, gilt aber auch für die Population der CD8+-T-Zellen und für Zellen mit einem γδ-T-Zell-Antigenrezeptor. Als Typ1-T-Zellen werden deshalb CD4+- oder CD8+-Lymphozyten definiert, welche typischerweise Interferon-γ (IFN-γ), IL-2, und TNF-α sezernieren. In Analogie werden CD4+- oder CD8+-Lymphozyten, welche charakteristischerweise die Zytokine IL-4, IL-5, IL-6, IL-10 und IL-13 bilden, als Typ2-T-Zellen bezeichnet. Diese Klassifizierung spiegelt extreme Reaktionsmöglichkeiten der Zytokin-sezernierenden T-Zellen wider und sollte deshalb als eine gewisse Vereinfachung der realen Verhältnisse betrachtet werden. Zusätzlich können sowohl im Gewebe als auch im peripheren Blut T-Zellen mit einem Zytokinmuster nachgewiesen werden, das typisch sowohl für Typ1- als auch für Typ2-T-Zellen ist und die gelegentlich auch als Typ0-T-Zellen bezeichnet werden.
Die Unterschiede zwischen Typ1-T-Zellen und Typ2-T-Zellen wurden erstmals 1986 von Tim Mosmann beschrieben.[4]
Cytotoxische T-Zellen
- → Hauptartikel: Cytotoxische T-Zelle
Cytotoxische T-Zellen (CTL), auch CD8+-Zellen genannt (veraltete Bezeichnung: T-Killerzellen), tragen typischerweise CD8+-αβ-Heterodimere auf ihrer Oberfläche und erkennen Antigene, die ihnen von kernhaltigen Zellen auf MHC-I-Molekülen dargeboten werden. Sie spielen vor allem in der Erkennung und Beseitigung von viral infizierten Zellen und Tumorzellen eine Rolle. Sie sind in der Lage, in diesen Zellen über verschiedene Signalwege - (Fas/FasL; Perforin/Granzyme) - den programmierten Zelltod herbeizuführen.
Regulatorische T-Zellen (TReg)
- → Hauptartikel: Regulatorische T-Zelle
Immunantworten sind sowohl quantitativ als auch qualitativ darauf ausgelegt, eine optimale Abwehrleistung zu erzielen. Unterschiedlichste Krankheitserreger müssen erfolgreich bekämpft werden, auf der anderen Seite muss die natürliche Tendenz zur Autoimmunität unter Kontrolle gehalten werden. Außerdem ist die Lymphozyten-Effektorpopulationen, durch hämatopoetische Prozesse, in ausreichender Anzahl und Funktion bereitgestellt werden. Die Kontrollmechanismen, welche diesen Aufgaben zugrunde liegen, sind komplex und schließen für die Regulation autoreaktiver T-Zellen unterschiedliche Mechanismen ein. Hierzu gehören die Deletion von peripheren T-Zellen, die differentielle Wirkungsweise der Zytokine IL-10 und TGF-β, die Kompetition um Antigene, Wachstums- oder Differenzierungsfaktoren, die Limitierung der klonalen Expansion durch Aktivierung von CTLA4 sowie die Induktion des programmierten Zelltodes über Fas/FasL-vermittelte Signale.
In den letzten Jahren haben sich zusätzliche Hinweise gehäuft, dass auch so genannte regulatorische T-Zellen (ehemals „Suppressor-T-Zellen“ genannt) an der Limitierung einer Immunantwort gegenüber Fremdantigenen und an der Erhaltung der Toleranz gegenüber Selbst-Antigenen zentral beteiligt sein könnten. Aufgrund spezifischer Marker und spezieller Zytokinprofile können regulatorische T-Zellen sowohl phänotypisch als auch funktionell in unterschiedliche Subpopulationen (CD4+-CD25+-T-reg-Zellen, TR1-Zellen, TH3-Lymphozyten und NKT-Zellen, CD8+-regulatorische Zellen) unterteilt werden.
T-Gedächtniszellen
- → Hauptartikel: T-Gedächtniszelle
Als „immunologisches Gedächtnis“ sorgen T-Gedächtniszellen für einen verbesserten Schutz bei einer Re-Infektion. Als ehemalige effektive T-Zellen (TH1 oder TH2) sind sie in der Lage, die einmal ausgebildete spezifische Immunreaktion rasch wieder in Gang zu setzen. Sobald der Organismus wieder mit demselben Antigen in Kontakt kommt, lösen die T-Gedächtniszellen schnell und effektiv eine Abwehrreaktionen aus, indem sie sich erneut in Effektorzellen Umwandlung. Die nach einer Antigenstimulation erfolgte Aktivierung des adaptiven Immunsystems führt zu einer Expansion der T-Zellen um den Faktor 10 bis 100; einige von ihnen werden nach erfolgreicher Bekämpfung wiederum in Gedächtniszellen umgewandelt. Die Gedächtnis-Funktion kann sowohl durch CD4+ als auch durch CD8+-T-Gedächtniszellen übernommen werden.
NK-T-Zellen
Diese kleinere Population von zirkulierenden αβ-T-Zellen wird durch das an ihrer Oberfläche exprimierte Molekül NKR-P1A charakterisiert. Dieses Lektin-ähnliche Protein ist dem murinen Zellmarker NK1.1 vergleichbar und gilt als charakteristischer Marker für natürliche Killerzellen. Zusätzlich besitzen NK-T-Zellen noch andere, typischerweise von NK-Zellen exprimierte Marker einschließlich CD56, Neural cell adhesion moleclue-1 (NCAM-1) und CD57. In diesen Zellen können auch die zytotoxischen Effektormoleküle Perforin und Granzym nachgewiesen werden. Den NK-T-Zellen wird eine negativ-regulatorische Funktion bei der Kontrolle von Autoimmunerkrankungen zugesprochen.
γδ-Antigenrezeptor-positive T-Lymphozyten
- → Hauptartikel: γδ-Antigenrezeptor-positive T-Lymphozyten
Eine kleinere Population von T-Zellen zeichnet sich dadurch aus, dass sie einen Antigenrezeptor exprimieren, der sich aus den polymorphen γ- und δ-Ketten zusammensetzt. Morphologisch können einige der γδ-Antigenrezeptorpositiven Zellen als large granular leukocytes (LGL) ausgewiesen werden, da in ihrem Zytoplasma viele Granula vorkommen. Der Großteil dieser Zellpopulation hat keine CD4/CD8 Korezeptoren an ihrer Oberfläche.
Die γδ-Antigenrezeptor-positive T-Lymphozyten werden in die die Vδ1- und die Vδ2-Unterklasse aufgeteilt. [5]
T-Lymphozyten-gebundene Erkrankungen
Angeborene Immundefizienzen
Erbliche Immundefekte sind meist rezessiv und oft auf Mutationen in Genen auf dem X-Chromosom zurückzuführen. Genetische Defekte, die sowohl den T-Zell- wie auch den B-Zellarm der Immunantworten betreffen, werden unter dem Begriff schwere kombinierte Immundefekte (SCID) zusammengefasst. Durch diese Krankheit betroffene Kinder können nur in völlig keimfreier Umgebung oder nach Gabe von Antikörpern und nach erfolgreicher Knochenmarktransplantation überleben.
Beim Di-George-Syndrom können T-Zellen im Thymus nicht reifen, da kein normal entwickeltes Epithelgewebe im Thymus vorhanden ist, das die Reifung von Thymozyten in T-Lymphozyten reguliert.
Beim Nacktes-Lymphozyten-Syndrom fehlen den Patienten MHC-II-Moleküle auf Makrophagen und B-Zellen sowie auf dem Thymusepithel, was dazu führt, dass diese Personen einen Mangel an CD4+ T-Lymphozyten haben. Dieser Mangel führt dann gleichzeitig zu einem Antikörpermangel.
Erworbene Immundefizienzen
Erworbene Immundefekte können durch verschiedenen Krankheiten, durch Mangelernährung, durch schädlichen Effekte der Umwelt oder therapeutischer Maßnahmen verursacht werden.
Infektionen
Das Humanes Immundefizienz-Virus (HIV), infiziert CD4+ T-Lymphozyten, dendritische Zellen und Makrophagen, was zur Immunschwäche-Krankheit Aids führt. Die Viren HTLV I und HTLV II können bei Menschen und Primaten T-Lymphozyten befallen und verschiedene Erkrankungen auslösen, unter anderem die Adulte T-Zell-Leukämie und die Tropische Spastische Paraparese.
Allergische Reaktionen
T-Lymphozyten spielen bei Überempfindlichkeitsreaktionen eine Rolle. Von einer Überempfindlichkeitsreaktion spricht man, wenn eine unangemessene Immunrektion gegen körpereigenes Gewebe oder auf ein eigentlich harmloses Antigen (Staub, Pollen, Nahrungs- oder Arzneimittel) ausgelöst wird. Es werden vier Typen von Überempfindlichkeitsreaktionen unterschieden; besonders beim Typ-I (Soforttyp) und Typ-IV, spielen die T-Zellen eine wichtige Rolle. Die Typ-I Überempfindlichkeitsreaktion ist durch ein Übergewicht der T2-Antwort gekennzeichnet, hingegen wird der Typ-IV meist durch T1-Zellen vermittelt. Bei letzterem kommt es zu einer anhaltenden Stimulation der T2-Zellen in Form einer persistierenden Entzündung (z. B. Tuberkulose), die zu einer Schädigung der betroffenen Gewebe führt.
Autoimmunerkrankungen
Autoreaktive T-Zellen können Autoimmunerkrankungen hervorrufen, lassen sich jedoch nur schwer nachweisen. Bei Diabetes mellitus-Typ-I ist bekannt, dass Insulin-spezifische CD4+ T-Zellen für den Untergang von β-Zellen des Pankreas verantwortlich sind. Es gibt auch Hinweise auf eine Beteiligung von T-Zellen an der rheumatoiden Arthritis [6]. Die Immunantwort bei Multiple Sklerose wird, nach gegenwärtigem Erkenntnisstand, durch aktivierte autoreaktive T-Zellen eingeleitet. Dabei kommt es zur Schädigung der Myelinscheiden um die Axone von Nervenzellen im Gehirn und Rückenmark.
Den γδ-T-Zellen werden zusätzliche immunregulatorische Funktionen bei der Pathogenese der Kontakt-Hypersensitivität und bei der chronischen Dermatitis zugeschrieben.
Medikamentenwirkungen
Bestimmte Arzneimittel können erwünschte und unerwünschte Immundefizienzen hervorrufen.
Erwünschte Medikamentenwirkungen
Nach der Transplantation eines Organs (Leber, Lunge, Nieren, Herz) oder Gewebe ist die Gefahr einer Transplantabstoßung sehr hoch. Diese Abstoßungsreaktion ist ein komplexer Prozess, in welchem zelluläre wie auch humorale Immunreaktionen eine Rolle spielen. Die T-Zellen sind jedoch bei der Abstoßung von zentraler Bedeutung. Das Gewebe eines anderen Individuums wird auf Grund einer zellulären Reaktion gegen allogene und xenogene MHC-Moleküle abgestoßen. Antigene des Transplantats können direkt durch T-Zellen des Empfängers oder indirekt durch Antigenpräsentierende Zellen erkannt werden. Daraus kann die Abstoßung auf drei Wegen erfolgen: Die akute Abstoßung durch CD8+ T-Zellen, durch die Schädigung der Gefäße und die chronische Abstoßung, welche durch CD4+ T-Zellen erfolgt. Um die Abstoßung zu verhindern werden heute dauerhaft immunsuppressive Medikamente eingesetzt.
Unerwünschte Medikamentenwirkungen
Durch Medikamente wie Chemotherapeutika oder durch Bestrahlung können erworbene Immunschwächen verursacht werden. Von dieser Nebenwirkung sind oft auch die T-Lymphozyten betroffen.
Onkologische Krankheitsbilder
T-Zellen können für die Entstehung verschiedener Tumorerkrankungen verantwortlich sein. Nach der Kiel-Klassifizierung der Non-Hodgkin-Lymphome unterschiedet man folgende Gruppen hochmaligner Lymphome:
- Pleomorphe, klein, mittelgroß- und großzellige T-Zell-Lymphome
- Großzellig-anaplastisches Lymphom (Ki-1-Lymphom)
- Immunoblastisches T-Zell-Lymphom
- Lymphoblastisches T-Zell-Lymphom
- Intermediär maligne Lymphome (Angioimmunoblastisches T-Zell-Lymphom (AILD))
- Niedrig maligne Lymphomedie (Chronische lymphatische Leukämie vom T-Zelltyp)
- Kleinzellig-zerebriformes Lymphom (Mycosis fungoides, Sézary-Syndrom, Lymphoepitheliodzellige Lymphom, Lennert Lymphom)
Die Akute lymphatische Leukämie (ALL) ist eine bösartige Tumorerkrankung, die in ca 50% der Fälle Kinder betrifft. Bei etwa einem Viertel der Fälle entsteht die ALL aus T-Lymphozyten.
Eine chronische lymphatische Leukämie (CLL) ist ebenfalls ein Lymphom - also im Lymphsystem entstanden. Die Bezeichnung Leukämie (weißes Blut) ist auf die krankhafte Vermehrung weißer Blutkörperchen im Blut zurückzuführen. Das ist kein Widerspruch, da die Definitionen für Leukämien (weißes Blut) und Lymphome (von malignen Zellen des Lymphsystems ausgehend) verschiedene Aspekte ansprechen und sich deshalb nicht ausschließen. Die, im Vergleich zur B-Zell Variante aggressiver T-Zell-Form macht etwa 5 % aller Fälle aus.
Das Vorkommen der T-Lymphozyten in anderen Lebewesen
In Wirbellosen (wie Einzellern, Schwämmen, Ringelwürmern und Arthropoden) finden sich weder Lymphozyten noch Lymphknoten. In Wirbeltieren kommen Lymphknoten erst bei der Vögeln und Säugetieren vor, dagegen sind die Lymphozyten schon eher im Stammbaum bei Knorpel- und Knochenfische sowie Amphibien und Reptilien vorhanden.
Forschungsgeschichte
Die beiden letzten Jahrzehnte des 19. Jahrhunderts waren von den Auseinandersetzungen der beiden Lager der zellulären und der humoralen Immunität geprägt.
Von der Phagozytosenlehre bis zur Entwicklung der Hypothese der Antikörperbildung
 Ilja Metschnikoff (Vertreter der Phagozytosenlehre)
Ilja Metschnikoff (Vertreter der Phagozytosenlehre)Der Zoologe Elias Metschnikoff (1845–1916) untersuchte intrazelluläre Verdauungsvorgänge bei Coelenteraten. Dabei beobachtete er, dass sich um einen in einen Seestern gestochenen Rosendorn bewegliche Zellen ansammelten. Metschnikoff, ein Vertreter der Phagozytosenlehre, nahm daher an, dass die bakterielle Entzündung ein Heilungsvorgang des Körpers sei - ein Abwehrmechanismus, während dem die Bakterien ,von den an den Infektionsort wandernden Zellen, „aufgefressen“ werden. Der Schule der Phagozytenlehre stand die - vor allem von deutscher Seite vertretene - Vorstellung einer humoralen Immunität gegenüber.
Emil Adolf von Behring (1854–1917) hatte, 1888 festgestellt, dass das Blutserum, der für Milzbrand hochempfindlichen Meerschweinchen das Wachstum von Bacillus anthracis nicht behindert. Das Serum milzbrandresistenter Ratten dagegen ließ kein Wachstum des Bakteriums zu.
Immunseren von Meerschweinchen, die mit Vibrio metschnikovii immunisiert waren, töteten in vitro diese Keime ab. Das Normalserum dieser Tiere war dazu nicht in der Lage. Andere Keime, etwa Milzbrandbazillen, wurden durch das Vibrionen-Immunserum nicht beeinflusst. Diese Befunde haben die unter anderem von Hans Buchner vertretene Auffassung widerlegt, dass das Blutserum eine unspezifische bakterizide Aktivität habe. Die Spezifität der Immunität gegen verschiedene Vibrionen hatte Richard Pfeiffer bereits 1889 festgestellt. Das Ergebnis der Zusammenarbeit von Behring und Kitasato bildete die Grundlage der Lehre von der humoralen Immunität und die so genannte „Blutserumtherapie“.
 Emil von Behring (Vertreter der Theorie der Humoralen Immunität)
Emil von Behring (Vertreter der Theorie der Humoralen Immunität)Die Brücke zwischen den zueinander in schroffem Gegensatz stehenden Lehren von der humoralen und der zellulären Immunität wurde von zwei Arbeiten der Belgier Denys und Lecleff beziehungsweise Denys und Marchand 1895/1896 geschlagen. Erst Almroth Wright und S.R. Douglas (1903,1904) beendeten diesen Streit. Die von ihnen im Serum nachgewiesenen, phagozytosefördernden, wärmeempfindlichen Stoffe wurden als Opsonine bezeichnet (Antikörper, welche nach Anheftung an die Bakterien die Phagozytose durch Granulozyten fördern). Damit löste sich der Widerspruch zwischen humoraler und zellulärer Immunität auf, und zuvor gemachte Beobachtungen über lytische Substanzen im Blut fanden ihre Erklärung.
Die Instruktions- oder Matrizentheorie von Linus Pauling (1940) ging von der Annahme aus, dass das Antigen in der Zelle eine Matrize für den sich bildenden Antikörper sei, wobei ein universelles Immunoprotein in Gegenwart des Antigens als Matrize strukturell zu dem spezifischen Antikörper umgeformt werde.
Niels Jerne vertrat wieder eine reine Selektionstheorie, die der Ehrlichschen Seitenkettentheorie nahe steht. Danach wurde angenommen, dass sämtliche Immunoglobuline vorgeformt seien und sich mit einem eingeführten Antigen verbinden würden. Frank MacFarlane Burnet ging einen anderen Weg, er nahm im Gegensatz zu Jerne an, dass nicht natürliche Antikörper mit dem Antigen reagieren, sondern Zellen selektiert werden. Diese Theorie setzt voraus, dass während des embryonalen Lebens durch somatische Mutationen die Antigenrezeptoren der Zellen selektiert und gleichzeitig Zellen, die Rezeptoren für körpereigene Antigene besitzen, eliminiert werden. Bereits Ehrlich hatte auf den „Horror autotoxicus“ hingewiesen, bei dem keine Antikörper gegen körpereigene Antigene gebildet werden. Um das für die Antikörperbildung erforderliche Wechselspiel zwischen B- und T-Lymphozyten zu beschreiben, wurde von Jerne 1974 die Netzwerktheorie des Immunsystems aufgestellt (Nobelpreis 1984).
Die Geschichte der Entdeckung der T-Lymphozyten
Zwischen 1911 und 1926 wurden Tumorerkrankungen untersucht und dabei festgestellt, dass Lymphozyten wahrscheinlich eine Rolle bei der Abstoßung von körperfremden Gewebe spielen.[7][8] 1964 wurde demonstriert, dass kleine Lymphozyten kontinuierlich aus dem Milchbrustgang ins Blut und, über die sekundären Lymphorgane, dann wieder zum Milchbrustgang zurück zirkulieren[9]. 1968 ist die Rolle des Thymus bei der Leukämie von Mäusen entdeckt und beschrieben worden.[10] Mitte der 1960er Jahre wurde aufgeklärt, dass die Funktion der B- und T-Lymphozyten für die normale Immunität wichtig sind. 1975 wurde die phänotypische und strukturelle Separation von zytotoxischen und nicht-zytotoxischen T-Zellen festgelegt.[11][12] Im Gegensatz zum B-Zell-Antigenrezeptor wird der TCR nicht wie ein Antikörper sezerniert, deshalb dauerte es für die Immunologen länger, genügend Proteine für eine strukturelle Untersuchung zu gewinnen. Erst 1976 als Rolf Zinkernagel und Peter Doherty zeigten, dass die Aktivierung des T-Zell-Antigenrezeptors nicht nur die Bindung des Antigens, sondern des gleichzeitigen Erkennens des MHCs bedarf[13], wurde es erst im Jahre 1982 möglich einen mAb zu generieren, der eine spezielle Struktur auf T-Zell-Lymphomen bei Mäusen erkannte.[14] Nach diesem ersten Schritt wurden T-Zell-Klon spezifische Strukturen, T-Zell-Hybridomas und T-Zell-Leukämie-Zelllinien beschrieben[15][16] die alle dieselbe Struktur aufwiesen, und zwar die TCR-Struktur. 1979 hat Kung et al. die CD3-Proteine (damals unter der Name "T3 complex") auf der Zelloberfläche an der Seite des T-Zell-Antigenrezeptors entdeckt [17] deren biochemische Charakterisierung [18] erfolgte 1984 in der Forschungsgruppe von Cox Terhorst. Die erste biochemische und strukturelle Charakterisierung des TCRs hat das Protein als ein 45-50 kDa großes Heterodimer, mit einer α- und einer β-Kette beschrieben.[19][20] Die mRNA-Isolation des menschlichen TCRs folgte - ebenfalls im Jahr 1984 - der bei Mäusen. Damals wurde erstmals das Klonieren von β-Ketten des humanen und Maus-TCRs beschrieben[21][22]. Erst nach Experimenten mit Klonen der αβ-TCR-Gene wurde erkannt, dass ein zweiter, dem αβ-TCR ähnlichen strukturellen Charakter besitzender TCR existiert - der γδ-T-Zell-Antigenrezeptor[23]. In demselben Jahr wurde erkannt, dass der T-Zell-Antigenrezeptor – im Gegensatz zum B-Zell-Antigenrezeptor – ein Antigen nicht direkt, sondern nur durch Bindung an MHC erkennen kann[24]. TCR-Gene können in chromosomale Verschiebungen mit einbezogen werden, die bei lymphoiden Erkrankungen Onkogene aktivieren. Mit Hilfe molekularer Proben von TCR konnten Gene identifiziert werden, die bei der Entwicklung von Leukämien und Lymphomen eine Rolle spielen[25]. 1988–89 wurde gezeigt, dass CD8 der Erkennungspartner für Antigene ist, die zusammen MHC-I präsentiert werden. Außerdem erkannte man, dass das Gedächtnis von CD4- und CD8-Zellen selbst lange nach einer eventuell nur kurzen MHC-Bindung, auch in Abwesenheit des MHC, erhalten bleiben kann[26][27].
Literatur
- G. A. Holländer: Immunologie, Grundlagen für Klinik und Praxis. 1. Auflage. Elsevier, München 2006, ISBN 3-437-21301-6.
- M. J. Owen, J. R. Lamb: Immunerkennung. Thieme, Stuttgart 1991, ISBN 978-3137541011.
- I. Jahn: Geschichte der Biologie, Theorien, Methoden, Institutionen, Kurzbibliographien. 3. Auflage. Gustav Fischer, Jena 1998, ISBN 3-437-35010-2.
- A. Wollmar, T. Dingermann: Immunologie, Grundlagen und Wirkstoffe, unter Mitarbeit von I. Zündorf. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart 2005, ISBN 3-8047-2189-3.
- Bucher O., Wartenberg H.: Cytologie Histologie und mikroskopische Anatomie des Menschen. 11. vollständig überarbeitete Auflage Auflage. Verlag Hans Huber, Bern, Stuttgart, Toronto 1992, ISBN 3-456-81803-3.
- Munk K.: Grundstudium Biologie Zoologie. Gustav Fischer, Heidelberg, Berlin 2002, ISBN 3-8274-0908-X.
Einzelnachweise
- ↑ Y. Y. Kong, H. Yoshida, I. Sarosi, H. L. Tan, E. Timms, C. Capparelli, S. Morony, A. J. Oliveira-dos-Santos, G. Van, A. Itie, W. Khoo, A. Wakeham, C. R. Dunstan, D. L. Lacey, T. W. Mak, W. J. Boyle, J. M. Penninger: OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. In: Nature. 397, Nr. 6717, 28. Januar 1999, S. 315–323 (PMID 9950424).
- ↑ S. Cenci, M. N. Weitzmann, C. Roggia, N. Namba, D. Novack, J. Woodring, R. Pacifici: Estrogen deficiency induces bone loss by enhancing T-cell production of TNF-alpha. In: Journal of Clinical Investigation. 106, Nr. 10, November 2000, S. 1229–1237 (PMID 11086024).
- ↑ Girardi M.: Immunosurveillance and immunoregulation by γδ T cells. In: J. of Investigative Dermatology. Nr. 126, 2006, S. 25-31 (PMID 16417214).
- ↑ Tim Mosmann, H Cherwinski, M. W. Bond, M. A. Giedlin, R. L. Coffman: Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. In: Journal of Immunology. 136, Nr. 7, 1986, S. 2348–2357 (Abstract).
- ↑ Kabelitz D., Wesch D., He W.: Perspectives of γδ T cells in tumor immunology. In: Cancer Research. Nr. 67, 2007, S. 5-8 (PMID 17210676).
- ↑ György Nagy, Joanna M. Clark, Edit Buzas, Claire Gorman, Maria Pasztoi, Agnes Koncz, Andras Falus and Andrew P. Cope: Nitric oxide production of T lymphocytes is increased in rheumatoid arthritis. In: Immunology Letters. 118, Nr. 1, 2008, S. 55-8 ([1]).
- ↑ J.B.Murphy: Studies in tissue specifity: II. The ultimate fate of mammalian tissue implanted in chick embryo. In: Journal of Experimental Medicine. Nr. 19, 1914, S. 181-186.
- ↑ J.B.Murphy: Factors of resistance to heteroplastic tissue-grafting:studies in tissue specifity III. In: Journal of Experimental Medicine. Nr. 19, 1914, S. 513-522.
- ↑ J.L.Gowans, E.J.Knight: The route of re-circulation of lymphocytes in the rat. In: Proc.R.Soc.Lond.B.Biol.Sci.. Nr. 159, 1964, S. 257-282 (PMID 14114163).
- ↑ J.F.Miller: Immunological function of the thymus. In: Lancet. Nr. 2, 1968, S. 748-749.
- ↑ P.Kisielow, J.A.Hisrst, H.Shiku, P.C.Beverley, M.K.Hoffman, E.A.Boyse, H.F.Ottgen: Ly antigens as markers for functionally distinct subpopulations of thymus-derived lymphocytes of the mouse. In: Nature. Nr. 253, 1975, S. 219-220 (PMID 234178).
- ↑ H.Shiku, P.Kisielow, M.A. Bean, T.Takahashi, E.A.Boyse, H.F.Ottgen, L.J. Old: Expression of T-cell differentiation antigens on effector cells in cell-mediated cytotoxicity in vitro. In: Journal of Experimental Medicine. Nr. 141, 1975, S. 227-241 (PMID 1078839).
- ↑ R.M. Zinkernagel, P.C. Doherty: Restriction of in vitro T-cell mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. In: Nature. Nr. 248, 1974, S. 701-702 (PMID 4133807).
- ↑ J.P. Allison, B.W. McIntyre, D. Bloch: Tumor specific antigen of murine T-lymphoma defined with monoclonal antibody. In: J. Immunol.. Nr. 129, 1982, S. 2293–2300 (PMID 15661866).
- ↑ L.E.Samelson, R.N. Germain, R.H. Schwatz: Monoclonal antibodies against the antigen receptor on a cloned T-cell hybrid. In: Proc.Nat.Acad.Sci. USA. Nr. 80, 1983, S. 6972-6976 (PMID 6316339).
- ↑ R.D.Bigler, D.E.Fischer, C.Y.Wang, E.A.Kan, E.A. Rinnooy Kan, H.G.Kunkel: Idiotype-like molecules on cells of human T cell leukemia. In: J.Exp.Med.. Nr. 158, 1983, S. 1000–1005 (PMID 6604124).
- ↑ P. Kung, G.Goldstein, E. Reinherz, S.F. Schlossman: Monoclonal antibodies defining distinctive human T cell surface antigens. In: Science. 206, 1979, S. 347-349.
- ↑ H.C. Oettgen, J. Kappler, W.J.M. Tax, C. Terhorst: Characterisation of the two heavy chains of the T3 complex on the surface of human T lymphocytes. In: Journal of Biological Chemistry. 259, Nr. 19, 10. October 1984, S. 12039-12048 (PMID 6090452).
- ↑ O.Acuto, R.E:Hussey, K.A.Fitzgerald, J.P.Protentis, S.C.Meuer, S.F.Schlossman, E.L.Reinherz: The human T cell receptor:appearence in ontogeny and biochemical relationship of alpha and beta subunits on IL-2 dependent clones and T cell tumors. In: Cell. Nr. 34, 1983, S. 717-726 (PMID 6605197).
- ↑ J.Kappler, R.Kubo, K.Haskins, J.White, P.Marrack: The mouse T cell receptor: comparison of MHC-restricted receptors on two cell hybridomas. In: Cell. Nr. 34, 1983, S. 727-737 (PMID 6605198).
- ↑ Y.Yanagi, Y.Yoshikai, S.P.Clark, I.Aleksander, T.W.Mak: A human T cell-specific cDNA clone ancodes a protein having extensive homology to immunoglobulin chains. In: Nature. Nr. 308, 1984, S. 145-149 (PMID 6202421).
- ↑ S.M.Hedrick, D.I.Cohen, E.A.Nielsen, M.M.Davis: Isolation of cDNA clones encoding T cell specific membrane-associated proteins. In: Nature. Nr. 308, 1984, S. 149-153 (PMID 16116160).
- ↑ M.B.Brenner, J. McLean, D.P.Dialynas, J.L.Strominger, J.A. Smith, F.L.Owen, J.G. Seidman et.al.: Identification of a putative second T cell receptor. In: Nature. Nr. 322, 1986, S. 145-149 (PMID 3755221).
- ↑ Z.Dembic, W.Haas, S.Weiss, J.McCubrey, H.Kiefer, H. von Boehmer, M. Steinmetz: Transfer of specifity by murine alpha and beta T cell receptor genes. In: Nature. Nr. 320, 1986, S. 232-238 (PMID 2421164).
- ↑ W.H.Lewis, E.E:Michalopoulos, D.L.Williams, M.D.Minden, T.W.Mak: Breakpoints in the human T cell antigenreceptor alpha chainlocus in two T-cell leukemia patients with chromosomal translocation. In: Nature. Nr. 317, 1985, S. 544-546 (PMID 3876514).
- ↑ A.M.Norment, R.D.Salter, P. Parham, V.H. Engelhard, D.R. Littman: Cell-cell adhesion mediated by CD8 and MHC-I class I molecules. In: Nature. Nr. 336, 1988, S. 79-81 (PMID 3263576).
- ↑ D.Maspoust, V. Vezys, E.J.Wherry, R. Ahmed: A brief history of CD8 T cells. In: Eur. J. Immunol.. Nr. 37, 2007, S. 103-110 (PMID 17972353).
Weblinks




Wikimedia Foundation.