- Hirschsprungsche Krankheit
-
Klassifikation nach ICD-10 Q43.1 Hirschsprung-Krankheit ICD-10 online (WHO-Version 2006) Beim Morbus Hirschsprung (Megacolon congenitum) handelt es sich um eine Erkrankung des Dickdarms, die zur Gruppe der Aganglionosen gezählt wird.
Inhaltsverzeichnis
Name und Häufigkeit
Die Krankheit ist nach dem dänischen Pädiater Harald Hirschsprung benannt, der 1886 erstmals öffentlich über sie berichtete. Der Morbus Hirschsprung, auch unter dem Lemma Hirschsprungsche Krankheit in den Medizinlexika beschrieben, tritt durchschnittlich bei einem von 5000 Kindern auf, wobei Jungen prozentual gesehen deutlich häufiger betroffen sind als Mädchen.
Bei ca. 12 % der Säuglinge mit einem Down-Syndrom (Trisomie 21) kann Morbus Hirschsprung diagnostiziert werden. Kombinationen mit anderen Fehlbildungen (z. B. Mukoviszidose, Brachydaktylie) kommen vor - sie sind aber selten.
In 80 % der Fälle sind bei der Aganglionose nur das Rektum und/oder Sigma betroffen (Short-Segment-Aganglionose). Etwa 5 % gehören zur Long-Segment-Aganglionose, bei der der krankhaft veränderte Dickdarmabschnitt insgesamt 40 cm und mehr ausgedehnt ist. In weniger als 5 % der Fälle fehlen die Nervenzellen im gesamten Abschnitt des Dickdarmes, man spricht in diesem Fall von einem Jirasek-Zuelzer-Wilson-Syndrom. In einigen Fällen fehlen die Nervenzellen bis in den Dünndarm.
Ursachen
Ein Mangel an Ganglienzellen („Aganglionose“) im Bereich des Plexus submucosus bzw. myentericus (Auerbach-Plexus) führt zu einer Hyperplasie der vorgeschalteten Nervenzellen mit vermehrter Acetylcholin-Ausschüttung. Durch diese permanente Stimulation der Ringmuskulatur kommt es zu einem dauerhaften Zusammenziehen des betroffenen Darmabschnittes. Das übermäßig gebildete Acetylcholin wird durch eine kompensatorisch vermehrt produzierte Acetylcholinesterase abgebaut.
Späte Defekte der Neuroblasteneinwanderung, Reifungsstörungen eingewanderter Neuroblasten, temporäre Ischämien des Darms oder virale Infektionen beim Embryo kommen ursächlich in Frage. Es existieren Untersuchungen, die beim Morbus Hirschsprung Mutationen im sogenannten Ret-Protoonkogen und des Endothelinrezeptor-Gens (EDNRB) belegen.
Die Multiple endokrine Neoplasie Typ IIb kann mit der Symptomatik des M. Hirschsprung assoziiert sein. Auch dort liegt eine Mutation des Ret-Protoonkogens vor.
Auch da Heiraten unter Verwandten häufiger vorkommen, ist diese Krankheit bei den Amischen (USA) relativ häufig.
Auswirkung
Durch die Überstimulation der Ringmuskulatur entsteht eine Obstruktion (Stenose) - das betroffene Darmsegment, meist das Rektum, ist somit eingeengt. Die Darmentleerung kann nicht mehr regelgerecht erfolgen, wodurch eine schwere Obstipation (Verstopfung) entsteht. Vor dem verengten Segment erweitert sich dann durch Kotstauung das Darmlumen (Dilatation) und es kommt zum Megacolon. Dies wiederum führt zu Beschwerden wie Meteorismus (Blähungen) und Erbrechen. Durch die Ansammlung von Kot kann es zu einer Stuhlinkontinenz im Sinne einer „Überlaufenkopresis“ kommen. Bei etwa 15 % der Patienten weist das Megacolon unterschiedlich schwere und teilweise auch nekrotisierende Entzündungen durch Clostridium difficile auf.
Klinische Zeichen
Erste Hinweise geben die genannten Beschwerden, die in der Regel innerhalb der ersten Tage nach Geburt auffallen. Fehlender Mekoniumabgang („Kindspech“) oder ein Mekonium-Ileus (ansonsten typisch für Mukoviszidose) können auf Morbus Hirschsprung hinweisen.
Bei der rektalen Untersuchung zeigt sich eine leere Ampulle sowie ein enger Analkanal.
Bei Erwachsenen tritt Morbus Hirschsprung selten auf und fällt durch chronische Verstopfung auf. Der Darmabschnitt, in dem Nervenzellen fehlen, ist hier meist sehr kurz, weshalb die Diagnose eines Morbus Hirschsprung erst spät gestellt wird.
Diagnostik
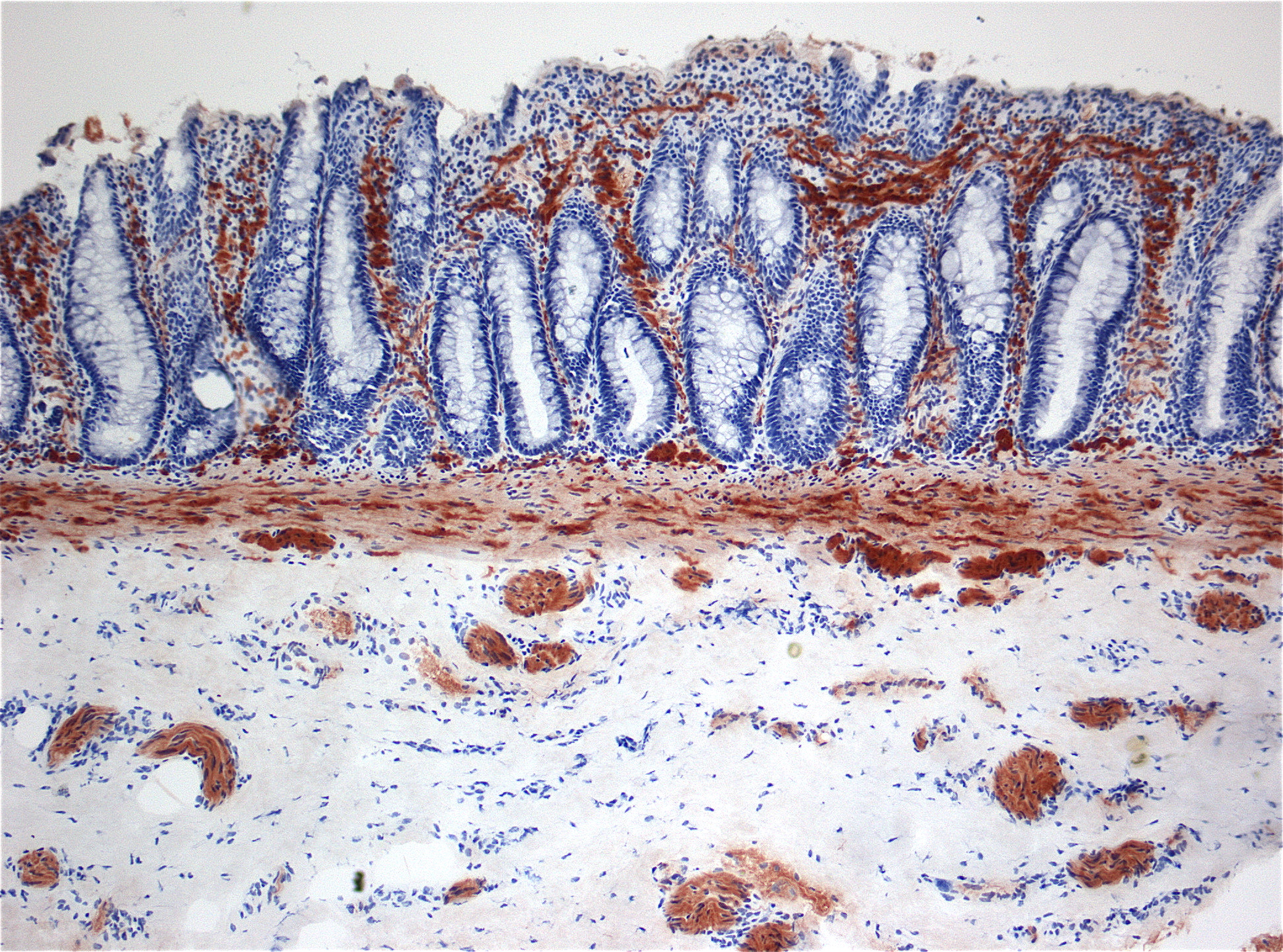
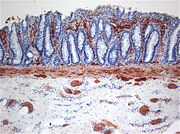 Fehlinnervation oberer Schleimhautschichten (Lamina propria) des Darms in der enzymhistochemischen Untersuchung der Acetylcholinesteraseaktivität (Braunfärbung).
Fehlinnervation oberer Schleimhautschichten (Lamina propria) des Darms in der enzymhistochemischen Untersuchung der Acetylcholinesteraseaktivität (Braunfärbung).Die Diagnose kann in der Regel radiologisch mittels eines Kolon-Kontrastmitteleinlaufs und mittels einer anorektalen Manometrie gestellt werden. Zur Diagnosesicherung wird unter Vollnarkose oder in Sedierung eine Saugbiopsie aus der Mastdarmschleimhaut vorgenommen. Bei ausreichender Biopsattiefe kann die pathologische Untersuchung in der NADH-Reaktion das Fehlen von Ganglienzellen bestätigen. Histochemisch lässt sich anhand der Acetylcholinesteraseaktivität nach den ersten Lebensmonaten eine cholinerge Fehlinnervation oberer Schleimhautschichten (Lamina propria) nachweisen.
Behandlung
Bei Neugeborenen wird bis zu einem operativen Eingriff meist vorübergehend ein künstlicher Darmausgang angelegt oder der Darm regelmäßig nach Anleitung gespült bzw. mit sogenannten Darmrohren so gut wie möglich geleert. Dieses wird in den After eingeschoben. Das Darmrohr wird derzeit aber immer seltener verwendet.
Als Therapie wird der betroffene Darmabschnitt operativ entfernt, bei sehr kurzem aganglionären Segment kann auch eine Sphinktermyektomie (Einschnitt des dauerkontrahierten Schließmuskels) durchgeführt werden. Als Operationsmethoden kommen je nach Erfahrung der Klinik offene, laparoskopische und transanale Methoden zum Einsatz.
Komplikationen
Bei unbehandeltem M. Hirschsprung: Enterocolitis, Peritonitis, Endotoxinschock (Sepsis durch von Bakterien erzeugte Toxine).
Postoperativ: weiterhin Obstipation/Diarrhoe
Quellen
Leitlinie der Deutschen Gesellschaft für Kinderchirurgie zum M. Hirschsprung
- Garcia-Barcelo M, Sham MH, Lee WS, Lui VC, Chen BL, Wong KK, Wong JS, Tam PK: Highly recurrent RET mutations and novel mutations in genes of the receptor tyrosine kinase and endothelin receptor B pathways in Chinese patients with sporadic Hirschsprung disease. In: Clin Chem. 2004 Jan;50(1):93-100. Epub 2003 Nov 18. PMID: 14633923, Volltext kostenlos bei NCBI
Weblinks
- Morbus Hirschsprung: Uniklinik Jena sehr verständliche, gut illustrierte Beschreibung (Uniklinik Jena)
- Die Chirurgie des Morbus Hirschsprung im Wandel der Zeit auf KidsDoc.at
Bitte beachte den Hinweis zu Gesundheitsthemen!
Wikimedia Foundation.