- 2D-Elektrophorese
-
 2D-Gel (Autoradiogramm)
2D-Gel (Autoradiogramm) 2D-Gele (Coomassie gefärbt)
2D-Gele (Coomassie gefärbt)Die zweidimensionale Gelelektrophorese oder 2D-Gelelektrophorese ist eine analytische Methode in Biochemie, Molekularbiologie und Proteomik. Sie wurde 1975 durch O'Farrell[1] und Klose[2] unabhängig voneinander entwickelt und kombiniert die isoelektrische Fokussierung (IEF) mit der SDS-Polyacrylamidgelelektrophorese (SDS-PAGE) zur Trennung komplexer Proteingemische (Bakterienlysate, Lysate von höheren Zellen oder Geweben, Körperflüssigkeiten) in Einzelproteine. Durch die Kombination der beiden orthogonal zueinander ausgeführten Trenntechniken wird eine besonders hochauflösende Trennung erreicht.
Jeder Fleck (Spot) im Proteinmuster entspricht einer Sorte (Spezies) von Proteinmolekülen. Da sich Proteinmuster in biologischen Systemen umwelt- und zustandsabhängig verändern, können sie zur Unterscheidung kranker und gesunder oder auch optimal und suboptimal gewachsener Zellen herangezogen werden. Sie geben beispielsweise Aufschluss über Krankheitsursachen oder den Wirkungsmechanismus von Medikamenten auf molekularer Ebene. Aufgrund der Komplexität von zweidimensionalen Proteinmustern wird für deren Auswertung auf speziell entwickelte Computerprogramme zurückgegriffen.
Probenvorbereitung
Um absolute Vergleichbarkeit der Proben zu gewährleisten, wird bei der Probenahme auf immer identische Bedingungen geachtet. Zur Gewinnung der Proteinextrakte werden die Zellen möglichst schonend geöffnet. Außerhalb der Zellstrukturen sind die austretenden Proteine besonders anfällig für die Bildung von Aggregaten und den Abbau durch ebenfalls in den Zellen vorhandene Proteasen. Da man im zweidimensionalen Muster Artefakte aus der Präparation vermeiden möchte, wird bei der Probenvorbereitung nahe 0 °C gearbeitet. Des Weiteren werden in der Regel Harnstoff und nichtionische Detergentien sowie proteasehemmende Stoffe zugesetzt.
Erste Dimension (IEF)
Bei der Isoelektrofokussierung (erste Dimension) werden die Proteine aus dem zu untersuchenden Extrakt auf der Basis ihres relativen Gehalts saurer und basischer Aminosäurereste aufgetrennt (Separation nach dem isoelektrischen Punkt des jeweiligen Proteins). Es stehen zwei verschiedene Isoelektrofokussierungstechnologien zur Verfügung.
- Es können immobilisierte pH-Gradienten (IPG) benutzt werden, die in einem Gelstreifen fixiert wurden. Der Gelstreifen besteht aus einer Polyacrylamidmatrix. Auf der einen Seite des Geles sind der Matrix Acrylamidderivate mit sauren, auf der anderen Seite des Gels mit alkalischen funktionellen Gruppen zugesetzt. Durch die Einpolymerisation in die Matrix kann sich der pH-Gradient nachträglich nicht mehr verändern.
- Weiterhin können sogenannte Trägerampholyte verwendet werden, die frei beweglich in einem Gelstreifen oder einem langen zylindrischen Gel (meist in einem Röhrchen) vorliegen und beim Anlegen eines elektrischen Feldes einen pH-Gradienten formen, der allerdings zeitinstabil ist.
Proteine sind über den größten Teil des pH-Spektrums geladen. Nur an einem genau definierten pH-Wert heben sich die Ladungen der basischen und sauren Aminosäuren eines Proteinmoleküls auf, so dass sich das Molekül nach außen ladungsneutral verhält. Es besteht also das gleiche Verhältnis von positiven zu negativen Ladungen, weswegen dieser Zustand auch als isoelektrischer Punkt (pI) bezeichnet wird (Nettoladung = +/-0).
 leeres IPG-Gel
leeres IPG-Gel mit Proteinen beladenes IPG-Gel
mit Proteinen beladenes IPG-Gel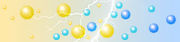 IPG-Gel während der IEF
IPG-Gel während der IEF IPG-Gel nach der IEF
IPG-Gel nach der IEF fertiges IPG-Gel
fertiges IPG-GelLegt man an den pH-Gradienten des mit Proteinen beladenen Isoelektrofokussierungsgels ein elektrisches Feld an, so wirkt auf alle geladenen Proteinmoleküle eine Kraft, die sie zu einer der beiden Elektroden zieht bzw. von ihr wegschiebt. Dabei durchlaufen die Proteine verschiedene Bereiche des pH-Gradienten. Wird genau der pH-Wert erreicht, der dem isoelektrischen Punkt des Proteins entspricht, so sind keine nach außen wirkenden Ladungen mehr vorhanden. Die Kraft des elektrischen Feldes kann nicht mehr auf das Protein wirken. Das Protein lagert sich ab. Entfernt sich das Protein dennoch durch Diffusion von dieser Position, erfolgt sofort eine Ladungsveränderung (durch veränderten Umgebungs-pH) und das elektrische Feld bringt es zurück an den pH-Wert, der dem pI des Proteins entspricht.
Äquilibrierung
Bei der sogenannten Äquilibrierung, die sich der Trennung nach dem pI anschließt, wird das Gel mit den Proteinen vorerst reduziert (beispielsweise mit Mercaptoethanol oder Dithiothreitol zur Beseitigung von Disulfidbrücken). Um eine Reoxidation der -SH HS-Gruppen zu -S-S-Gruppen zu verhindern, werden die HS-Gruppen z.B. mit Iodacetamid alkyliert. Im folgenden Schritt werden die Proteine mit Natriumdodecylsulfat (sodium dodecyl sulphate – SDS) beladen. SDS ist ein negativ geladenes Detergenz und heftet sich mit dem aliphatischen Ende an die Proteinmoleküle, mit der negativ geladenen Seite stößt es sich von in der Nachbarschaft gebundenen ebenfalls negativen SDS Molekülen ab, was zur völligen Entfaltung (Linearisierung) der Proteinmoleküle führt. Je größer ein Proteinmolekül, desto länger die entstehenden mit SDS beladenen Ketten. Da mehrere hundert negativ geladene SDS Moleküle an die Proteinmoleküle binden, kann die Eigenladung der Proteine im weiteren vernachlässigt werden.
Zweite Dimension (SDS-PAGE)
Den Gelstreifen mit den nach dem pH-Wert aufgetrennten und äquilibrierten Proteinen legt man auf die Kante eines quadratischen bzw. rechteckigen ebenfalls SDS-haltigen Polyacrylamidgels und trennt die Proteine nun senkrecht zur ersten Dimension in einer zweiten Elektrophorese nach ihrer Größe. Beim Anlegen des elektrischen Feldes (Anode gegenüber vom IEF-Gelstreifen) wandern die entfalteten und von SDS umgebenen Proteine mit ihrem Überschuss negativer Ladungen durch das Gel, welches den Proteinen entsprechend ihrer Molekülgröße einen mehr oder minder großen Widerstand entgegensetzt. Kleine Moleküle wandern relativ ungestört und erreichen schnell die dem IEF-Gel abgewandte Gelkante, große Moleküle werden bei der Wanderung ständig vom Gel gebremst und kommen kaum voran. Die Trennung in der zweiten Dimension wird mit Ankunft der kleinen Proteine am dem IEF-Gel abgewandten Gelrand gestoppt. Sichtbar wird dies durch einen mitlaufenden Farbstoff, wie zum Beispiel Bromphenolblau. Damit das Proteinmuster nach erfolgter Trennung vorhanden bleibt, muss es in einem abschließenden Schritt fixiert werden. Dazu werden Methanol und Essigsäure genutzt. Diese denaturieren die getrennten Proteine und verbinden sie mit der Gelmatrix. Dadurch wird Diffusion verhindert und das 2D Muster wird zeitstabil.

Leeres SDS-Gel

Gestartetes SDS-Gel
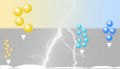
SDS-Gel während der Elektrophorese
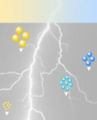
etwas später

Fertiges SDS-Gel}
Die Proteine markieren und detektieren
Markierung in der lebenden Zelle (in vivo)
Parameter wie die Produktion von Proteinen in einem bestimmten Zeitraum (Proteinsyntheserate) oder die Phosphorylierung von Proteinen pro Zeiteinheit lassen sich ausschließlich durch den Einbau von (radioaktiven) Isotopen bestimmen. Dazu wird der Bakterien/Zellkultur ein Nährstoff mit einem außergewöhnlichen Isotop verabreicht, welches dann in die Proteine (35S) oder in die Phosphatgruppen phosphorylierter (32/33P) Proteine eingebaut wird. Wird die Zeitspanne des Einbaus relativ kurz gewählt, ist im Ergebnis eher eine Momentaufnahme des zellulären Geschehens erfassbar, bei langer Zeitspanne eher das kumulierte Bild vieler Einzelereignisse. Der Proteinextrakt der markierten Zellen wird separiert und der Anteil der markierten Proteine über Autoradiographie oder Massenspektrometrische Verfahren im 2D Muster bestimmt. Zur Bestimmung der akkumulierten Proteinmenge kann neben einer dauernden Isotopenmarkierung auch auf eines der folgend erklärten Verfahren zurückgegriffen werden.
Markierung vor der gelelektrophoretischen Trennung
Besonders um mehrere Proben auf einem 2D Gel zu trennen, werden kovalent bindende Fluoreszenzfarbstoffe eingesetzt. Dazu werden maximal drei verschiedene Proteinextrakte mit je einem Farbstoff markiert, gemischt und gemeinsam auf demselben Gel getrennt. Da die Farbstoffe separat voneinander detekiert werden können, ist die Generierung und der differentielle Vergleich dreier probenspezifischer Proteinmuster möglich (DIGE - differential gel electrophoresis). Problematisch ist die Massenbeeinflussung der Proteine durch den gebundenen Farbstoff. Zudem sind die Farbstoffe relativ instabil und teuer.
Verwendet werden:
- Cy2; Cy3; Cy5
- Flashpro Farbstoffe
Markierung bzw. Färbung nach der Trennung
Die klassischen Proteinfärbungen erfolgen nach der elektrophoretischen Trennung. Je nach gewünschter Spezifität und Sensitivität werden eingesetzt:
Absorptionsfarbstoffe
- Coomassie-Brillant-Blau - Proteinmenge / nicht linear
- colloidal Coomassie-Brillant-Blau - Proteinmenge / nicht linear
- Silberfärbung - Proteinmenge / nicht linear / hoch sensitiv
- negative Zink Färbung - Proteinmenge / nicht linear / wenig sensitiv
Fluoreszenzfarbstoffe
- SyproRuby - Proteinmenge / linear / sensitiv
- Krypton-Färbung - Proteinmenge
- Flamingo-Färbung - Proteinmenge / linear / hoch sensitiv
- DeepPurple-Färbung - Proteinmenge
- Diamond ProQ - phosphorylierte Proteine (s. auch Posttranslationale Modifikation)
- Emerald ProQ - glycosylierte Proteine (s. auch Posttranslationale Modifikation)
Durch die zweidimensionale Gelelektrophorese lassen sich in bakteriellen Extrakten nach Färbung der Proteine oft weit über tausend verschiedene Proteinspezies nachweisen. In Mausembryonen konnte Klose ca. 10000 Spots darstellen.
2D Gele analysieren und interpretieren
2D Gele digitalisieren
Beim Digitalisieren von 2D-Gelen werden die 2D-Muster in Pixel mit verschiedenen Grauwerten zerlegt. Die Auflösung bestimmt die Genauigkeit in x- und y-Richtung, die Farbtiefe die Menge der Graustufen, welche für die Abbildung der Proteinmenge pro Pixel zur Verfügung steht.
Imaging-Geräte
- Für alle sichtbaren Absorptionsfarbstoffe werden höherwertige Weißlichtscanner mit hohem Probendurchdringungsvermögen (Durchlichtscans mit einer OD bis 4.0) eingesetzt. Ohne eine entsprechende Kalibrierung der Proteinkonzentrationen, Farbstoffsignale und Grauwerte sind quantitative Aussagen jedoch später nur bedingt möglich. Ebenfalls verfügbar für das Gele digitalisieren mit sichtbaren Farbstoffen sind inzwischen kamerabasierte Systeme. Farbfilter können u.U. die Ausnutzung des dynamischen Bereiches verbessern helfen.
- Zur Detektion von Fluoreszenzfarbstoffen werden hauptsächlich lichtstarke Laserscanner genutzt, bei denen die Wellenlänge des Anregungslichtes und die Filteroptik für das Abstrahlungslicht der Fluoreszenzfarbstoffe flexibel angepasst werden können. Auch hier werden die etablierten Systeme inzwischen durch kamerabasierte Geräte komplementiert.
- Für den Nachweis von radioaktiven Isotopen, welche in die Proteine eingebaut worden sind, werden sogenannte Phosphorimager eingesetzt. Hier wird eine sogenannte Imaging-Platte dem getrockneten 2D-Gel für mehrere Stunden ausgesetzt. Die freiwerdende radioaktive Strahlung wird in der Imaging-Platte gespeichert und vom Phosphorimager ausgelesen. Die dabei aus der Imaging-Platte freiwerdende gespeicherte Energie wird linear in Graustufen des digitalisierten Bildes übersetzt (s. Autoradiogramm oben).
- Ein gerade in den Markt eingeführtes System detektiert die Eigenfluoreszenz von Proteinen unter UV-Licht durch Anregung aromatischer Aminosäuren wie Tyrosin, Phenylalanin und Tryptophan. Diese neue Technologie erspart den Färbungsschritt. Dies führt zu massiver Zeitersparnis und zur Minderung von Diffusions- und anderen Effekten, die durch Färbe- und Waschschritte bedingt werden. Die Technologie steckt allerdings noch in den Kinderschuhen und ist nur bedingt einsetzbar.
Für weitere Informationen zum Scannen von Gelen kann man eine entsprechende Anleitung (in Englisch) herunterladen[3].
Single Channel-Techniken
Bei den Single Channel Techniken wird in der Regel eine Serie von Gelen aufgenommen, die vollständig auf die gleiche Art und Weise eingefärbt wurde.
Multiplexing
Beim Gel-Multiplexing werden von ein- und demselben Gel mehrere Bilder unabhängig voneinander generiert. Das kann unter verschiedenen Umständen möglich sein:
- Es werden verschiedene Farbstoffe oder Labels eingesetzt, die verschiedene Eigenschaften der getrennten Proteine darstellen.
- Beispielsweise können die Proteinmenge durch den Flamingofarbstoff und die Proteinphosphorylierung durch den ProQ-Diamond Farbstoff im selben Gel detektiert werden. Beide Fluoreszenzfarbstoffe werden separat vom Scanner detektiert und in zwei Graustufenbildern gespeichert. Diese können mit einer entsprechenden Imagingsoftware zu einem Falschfarbenbild zusammengesetzt werden.
-
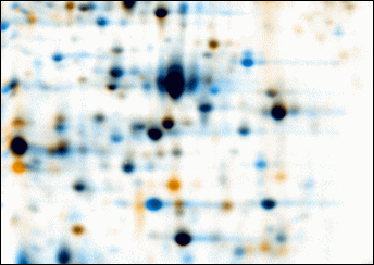
Eine weitere Möglichkeit ist die Kombination eines Autoradiogramms (dieses stellt die Proteinsynthese ermittelt durch eine radioaktive Pulsmarkierung dar) und eines Proteinmengenbildes (Detektion der Proteine mit der Silberfärbung). Im dargestellten Falschfarbenbild sind in rot die neu synthetisierten und in grün die akkumulierten Proteine sichtbar.
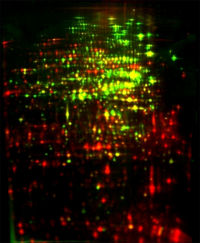 Visualisierung von Proteinmenge (grün) und Proteinsynthese (rot) (Software:Delta2D)
Visualisierung von Proteinmenge (grün) und Proteinsynthese (rot) (Software:Delta2D) - Kombinationen von Emerald ProQ Bildern (Färbung von Proteinen mit Zuckermolekülen als Seitenketten), Diamond ProQ Bildern (Färbung der Phosphatreste am Protein) und SyproRuby (Färbung der Proteinmenge) in gleichzeitig drei Farbkanälen kann gleichzeitig Phosphorylierung und Glykosylierung von Proteinen detektieren.
- Alternativ zur unabhängigen Detektion von verschiedenen Proteineigenschaften über verschieden spezifische Farbstoffe bzw. Labels können bei der DIGE bis zu drei Proben gleichzeitig auf einem Gel detektiert werden. Dazu werden die Proteine der drei Proben separat vor dem elektrophoretischen Lauf kovalent mit den Cyanin-Farbstoffen Cy2, Cy3 und Cy5 verknüpft. Danach werden die Proben zusammengefasst und simultan auf dem gleichen Gel getrennt. Die Detektion der Gele erfolgt mit drei Lasern verschiedener Wellenlänge und mit entsprechender Detektionsoptik.
2D Proteinmuster auswerten
Klassischerweise werden 2D-Gele visuell am Licht- oder bei Fluoreszenzfarbstoffen am UV-Tisch ausgewertet. Aufgrund der Komplexität von Proteinmustern führt eine softwaregestützte Analyse zu verlässlicheren Ergebnissen. Für einen Überblick über den aktuellen Stand der Vorgehensweise siehe [4] und [5].
Gelbilder aufbereiten
 In moderner Analysesoftware wird vom Originalbild Background und Artefaktbild abgezogen. Übrig bleibt das bereinigte Spotmuster, welches quantitativ ausgewertet werden kann
In moderner Analysesoftware wird vom Originalbild Background und Artefaktbild abgezogen. Übrig bleibt das bereinigte Spotmuster, welches quantitativ ausgewertet werden kannUm Gelbilder quantitativ zu analysieren, sollten sie vom 2D-Gel-typischen inhomogenen Hintergrund befreit und von artifiziellen Signalen bereinigt werden. Das nebenstehende Bild zeigt beispielsweise die Zerlegung eines Gelbildes in eine Hintergrundkomponente, eine Komponente mit artifiziellen Signalen und die für die weiterführende quantitative Analyse genutzte Spot-Komponente.
Gelbilder positionell korrigieren
Ein lange Zeit ungelöstes Problem stellte die schwierige positionelle Reproduzierbarkeit der Proteinmuster im 2D Gel dar. Eine mögliche Lösung bestand in der Vermeidung unabhängig hergestellter Gele.
- Die DIGE mit der simultanen Trennung von bis zu drei Proben pro Gel ist ein gangbarer Weg, wird allerdings bei mehr als drei Proben wieder mit dem Reproduzierbarkeitsproblem konfrontiert, da dann wiederum mehrere Gele miteinander verglichen werden müssen. Da es bis heute keine befriedigende experimentelle Lösung zur Vermeidung von Laufunterschieden gibt, musste auf Ebene der Softwareanalytik eine Lösung gefunden werden.
- Beim Spot Pattern Matching wird mit Hilfe einer Software im rohen 2D-Gelbild im ersten Schritt nach Proteinspots gesucht. Mit Hilfe der computergenerierten Spot-Information wird versucht, zu einem Expressionsprofil gehörende Partner zu finden (Spot Matching). Schwächen bei der Spotdetektion führen jedoch zu Spotzuordnungsfehlern bei diesen Verfahren.
- Das sogenannte Image Warping (Bildver- bzw. -entzerrung) wurde im Jahr 2000 in die 2D-Gel-Analyse eingeführt. Automatische Verfahren des Image Warpings nutzen die gesamte in den 2D-Gel-Bildern vorhandene Pixel-Information für die Berechnung einer Bildtransformation, die zur bestmöglichen positionellen Übereinstimmung der zu vergleichenden Gelbilder führt. Durch die positionelle Korrektur ist ein aufwendiges Spotmatching nicht mehr nötig, da zueinander gehörige Spots bereits an den gleichen Positionen der zu vergleichenden Gele liegen. Positionell transformierte Gele entsprechen sozusagen idealen Gelen, die keine systembedingten Verzerrungen mehr aufweisen.
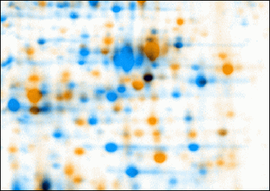 Image Warping: Zwei Gelbilder wurden in einem Falschfarbenbild kombiniert, das erste Bild orange, das zweite blau. Wegen der Laufunterschiede befinden sich einander entsprechende Spots nicht an der gleichen Position
Image Warping: Zwei Gelbilder wurden in einem Falschfarbenbild kombiniert, das erste Bild orange, das zweite blau. Wegen der Laufunterschiede befinden sich einander entsprechende Spots nicht an der gleichen Position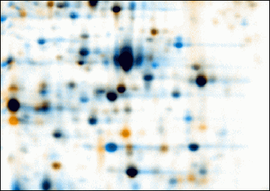 Image Warping: Die gleichen Bilder im Falschfarbenbild und transformiert. Einander entsprechende Spots befinden sich nun an der gleichen Position. Gemeinsame Spots in beiden Mustern sind schwarz, orange Spots sind nur im ersten Bild (verstärkt) vorhanden, blaue Spots nur im zweiten Bild.
Image Warping: Die gleichen Bilder im Falschfarbenbild und transformiert. Einander entsprechende Spots befinden sich nun an der gleichen Position. Gemeinsame Spots in beiden Mustern sind schwarz, orange Spots sind nur im ersten Bild (verstärkt) vorhanden, blaue Spots nur im zweiten Bild.
Referenzgele und Proteomkarten
Für eine wissenschaftlich fundierte Interpretation der 2D-Gele wird die Identität der hinter den Proteinspots stehenden Proteine bestimmt. Dies kann über verschiedene Technologien wie beispielsweise den Edman-Abbau oder massenspektrometrische Verfahren wie MALDI-TOF Massenspektrometrie erfolgen. Während in den 1990er Jahren die Proteinspots noch manuell aus den Gelen ausgeschnitten wurden, haben heute Roboter zum Ausstechen der Spots und zum Pipettieren in die Laboratorien Einzug gehalten. Mit Hilfe der Robotertechnik können mehrere hundert Proteine quasi über Nacht identifiziert werden.
 Für die Isolation von Proteinspots aus 2D-Gelen werden in modernen Labors Roboter zum Ausschneiden der Spots und zum Pipettieren eingesetzt.
Für die Isolation von Proteinspots aus 2D-Gelen werden in modernen Labors Roboter zum Ausschneiden der Spots und zum Pipettieren eingesetzt.Aufgrund der Einführung der computergestützten positionellen Korrektur der Proteinmuster wurde es möglich, Proteinspotidentifikationen problemlos von einem Gel auf ein anderes zu übertragen, ohne nochmals die Spots identifizieren zu müssen. Ein 2D-Gel, welches den Proteinextrakt aus einer Zellkultur zeigt, bildet nur ein Subset aller möglichen Zellproteine ab. Erst Gelserien aus Zellkulturen, die unter verschiedenen Wachstumsbedingungen gezogen wurden, können die Gesamtzahl aller möglichen Proteine zeigen. Grund ist die differentielle Genexpression, die nur die Produktion momentan wichtiger Proteine erlaubt und die Synthese gerade nicht benötigter Proteine verhindert. Zur Konstruktion umfassender Proteomekarten, die einen Großteil aller möglichen Proteine enthalten, werden Gel-Einzelbilder positionell angeglichen und über Bildfusionsalgorithmen zu einem Kompositbild zusammengefasst. Das Kompositbild wird mit den Daten aus den Proteinidentifikationen kombiniert und kann dann als Referenz für die Interpretation weiterer Experimente genutzt werden.
Proteinspots detektieren und quantifizieren
Für eine (semi)quantitative Auswertung der 2D-Gele wird die Gesamt-Absorption (Absorptionsfarbstoffe), das Gesamt-Radiosignal (radioaktiv markierte Proteine, Autoradiogramm) bzw. das Gesamt-Fluoreszenzsignal (Fluoreszenzfarbstoffe) eines Proteinspots über alle Bildpunkte ermittelt. Im ersten Spotdetektionsschritt werden die Koordinaten der Proteinspots und im zweiten die entsprechenden Spotformen bestimmt. Die Bestimmung der Spotumrisse kann nah an der Pixelinformation oder aber über mathematische Modelle erfolgen. Störinformationen, wie z.B. Hintergrund, Artefakte und Bildrauschen (s. Bildvorbereitung) werden vor der Quantifizierung ausgeschlossen. Die Grauwerte der Bildpunkte werden wenn nötig mit geräte- und farbstoffabhängigen Kalibrierungskurven korrigiert und dann innerhalb der gefundenen Spotumrisse zu einer Rohquantität aufsummiert. Die Rohquantitäten werden normiert und den entsprechenden Proteinspots zugeordnet. Da wie weiter oben schon erwähnt die Spotdetektion von Gel zu Gel nicht absolut reproduzierbare Ergebnisse liefert, kann es beim Zuordnen von Proteinen zu ihren Expressionsprofilen zu Irrtümern kommen, die mit den etablierten Methoden manchmal nicht aufgelöst werden können. Darum wurde 2003 eine neue Methode zum Spotmatching eingeführt. Diese beruht auf der Definition eines Spotconsensus aus allen zu analysierenden Gelen eines Experiments auf der Basis eines Kompositbildes. Weil das Kompositbild sämtliche Spotinformationen aus dem Gesamtexperiment enthält, kann der Spotconsensus mindestens auf all jenen Gelen zur Spotquantifizierung angewendet werden, aus denen das Kompositbild erstellt wurde. Spotzuordnungsfehler können bei Anwendung dieser Methode ausgeschlossen werden.
Daten visualisieren
 Visualisierung der Proteinexpression durch Farbcodes (Software:Delta2D)
Visualisierung der Proteinexpression durch Farbcodes (Software:Delta2D)Durch die Anwendung von Proteomekarten und kompositbildbasierter Spotdetektion ergeben sich völlig neue Möglichkeiten der Visualisierung von Proteinspots, die im analysierten Experiment auffällig wurden. Das nebenstehende Bild beispielsweise zeigt eine Zusammenfassung aus vier verschiedenen Proteinsynthesemustern. Die Farben zeigen an, unter welchen Umweltbedingungen welches Protein in seiner Synthese mindestens zweifach erhöht wird. Mit Hilfe derartiger Farbcodierungen wird es erstmals möglich, neben den positionellen Daten der Proteinspots nun auch Regulationsdaten zu visualisieren. Biomarker sind somit schnell und zuverlässig identifizierbar.
Vorteile von 2D-Gelen
- Die Visualisierung des Proteinmusters erlaubt bereits ohne weitergehende Analyse eine qualitative Einschätzung der Ergebnisse
- Verschiedene Proteinspezies ein- und desselben Primärproteins sind ohne Vorbedingung einer Analyse sofort zugänglich
- Die Methode ist hoch parallelisierbar. Je nach 2D-Gelelektrophoresegerät können bis zu 12 Gele gleichzeitig produziert werden.
- Die nötigen Investitionen sind gegenüber gelfreien Methoden relativ überschaubar.
- Das separierte Gel kann längere Zeit gelagert und später weiterverarbeitet werden.
- Inzwischen stehen Analysesysteme zur Verfügung, die in kurzer Zeit und ohne erweiterte Computerkenntnisse eine Vielzahl von Proben vergleichen können.
Probleme der 2D-Gel-Elektrophorese
Wie jede andere Technik in der Proteinbiochemie birgt auch die 2D-Gel-Elektrophorese einige Probleme:
- Aufgrund des Trennsystems im wässrigen Milieu werden hauptsächlich hydrophile Proteine mit einem GRAVY Index kleiner und nahe 1 aufgetrennt.
- Trotz des großen Separationspotenzials können starke Proteinspots durchaus schwache Spots überlappen bzw. völlig maskieren. Diese maskierten Spots sind einer quantitativen Analyse nicht mehr zugänglich.
- Zu große Gesamtproteinmengen führen zu Diskriminierungsphänomenen beim Eintritt verschiedener Proteinspezies in das IEF-Gel und somit zu einer Verzerrung der Proteinspezies-Verhältnisse.
- Je basischer ein Protein ist, desto schwerer wird dessen Separation. Ferner ist das 2D-Gelsystem hauptsächlich in einem pH-Bereich von 3-10 effizient.
- Die 2D-Gel-Elektrophorese stellt höchste Ansprüche an Ausdauer, manuelles Geschick und Gewissenhaftigkeit des Experimentators.
Einzelnachweise
- ↑ O'Farrell, P. H.: High resolution two-dimensional electrophoresis of proteins.,J. Biol. Chem. 250, 1975, 4007-4021. PMID 236308; PDF (freier Volltextzugriff)
- ↑ Klose, J.: Protein mapping by combined isoelectric focusing and electrophoresis in mouse tissues. A novel approach to testing for induced point mutations in mammals.; Humangenetik 26, 1975, 231-243. PMID 1093965
- ↑ http://www.decodon.com/Support/Howto/Scanning/scanning_2D_gels.html Scanning Guide (Englisch)
- ↑ Berth M, Moser FM, Kolbe M, et al. (2007). The state of the art in the analysis of two-dimensional gel eletcrophoresis images. Appl Microbiol Biotechnol. 2007;76(6):1223–43. [1] (Dieser Artikel ist frei verfügbar unter der Springer Open Access Lizenz.)
- ↑ Bandow J, Baker JD, Berth M, Painter C, et al. (2008). Improved image analysis workflow for 2-D gels enables large-scale 2-D gel-based proteomics studies - COPD biomarker discovery study. Proteomics 2008 [2]
Literatur
- Allen, R.C., Budowle, B.: Gel Electrophoresis of Proteins and Nucleic Acids: Selected Techniques. Walter de Gruyter; 1994. ISBN 3-11-013896-4
- Bandow J, Baker JD, Berth M, Painter C, et al.: Improved image analysis workflow for 2-D gels enables large-scale 2-D gel-based proteomics studies - COPD biomarker discovery study. Proteomics 2008 [3]
- Berth M, Moser FM, Kolbe M, et al: The state of the art in the analysis of two-dimensional gel eletcrophoresis images. Appl Microbiol Biotechnol. 2007;76(6):1223–43. [4] (Dieser Artikel ist frei verfügbar unter der Springer Open Access Lizenz.)
- Hamdan, M.H., Righetti, P.G.: Proteomics Today: Protein Assessment and Biomarkers Using Mass Spectrometry, 2D Electrophoresis,and Microarray Technology. Wiley-Interscience; 2005. ISBN 0-471-64817-5
- Rabilloud, T. (Hrsg.): Proteome Research: Two-Dimensional Gel Electrophoresis and Identification Methods (Principles and Practice). Springer; 1999. ISBN 3-540-65792-4
- Schmitz, Sabine: Der Experimentator: Zellkultur. Spektrum Akademischer Verlag; 1. Auflage 2007. ISBN 3-8274-1564-0
Weblinks
- fixingproteomics.org Protocols for preparing samples and running 2-D gels (English).
- The state of the art in the analysis of two-dimensional gel electrophoresis images
Software zur Analyse und Auswertung von 2D-Gelen
- Delta2D
- JVirGel
- PDQuest
- Progenesis SameSpots
- Z3
(wird nicht mehr weiter entwickelt)










Wikimedia Foundation.